


Intervention Studies (Clinical Trials, Experimental Studies)
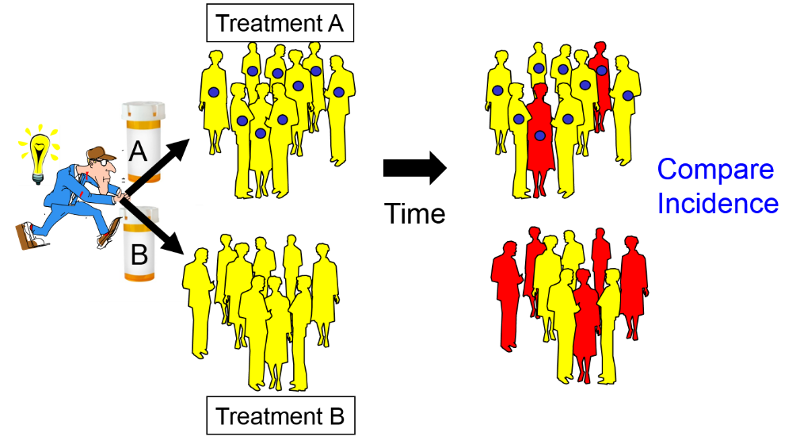
Intervention studies (clinical trials) are similar to prospective cohort studies in design in that subjects with or without a given exposure are followed over time to compare incidence of the outcome of interest. The key difference is that prospective cohort studies are observational, but in clinical trials the investigators assign subjects to the exposure groups
While this design is frequently used to evaluate new drugs, it can be used to evaluate the efficacy of
- Diets (e.g., primary prevention of cardiovascular disease with a Mediterranean diet)
- Exercise regimens (e.g., a clinical trial of exercise to alleviate post-partum depression)
- New programs (e.g., pre-natal care in groups of 8-10 women versus usual one-on-one pre-natal care)
- New clinical management schemes (e.g., a protocol to reduce post-operative complications)
However, unlike prospective cohort studies in which investigators record exposures that subjects already have, in clinical trials the investigators assign patients to one of the exposure groups being compared. Ideally, this assignment is done with random allocation, meaning that each subject has an equal chance of being assigned to any one of the "exposures."
Ethical Considerations
Investigators assign patients to competing treatments in clinical trials, and this raises the question of whether it is ethical to do this. Certainly, it is not ethical to test all exposures in this fashion. It would be unethical, for example, to conduct a clinical trial on the effects of smoking, particularly since we know that the harm caused by smoking far outweighs any potential benefits, such as relaxation or weight control.
On the other hand, consider a situation in which a new drug has been developed to treat breast cancer. Perhaps it has been found to be effective in cell cultures and in animal models, and perhaps preliminary studies in small groups of human volunteers have shown some evidence of effectiveness with minimal side effects. tIn other words, there is reason to believe that it might be a beneficial new treatment, but there is also doubt.about effectiveness and possible side effects. Testing on a large scale with a comparison group may show that it is not so effective or that its side effects are unacceptable. This is what is referred to as equipoise, i.e., the balance between sufficient belief in its potential benefit and safety that one can justify exposing some subjects to it and sufficient doubt about its benefit and safety that one can justify withholding it from some subjects.
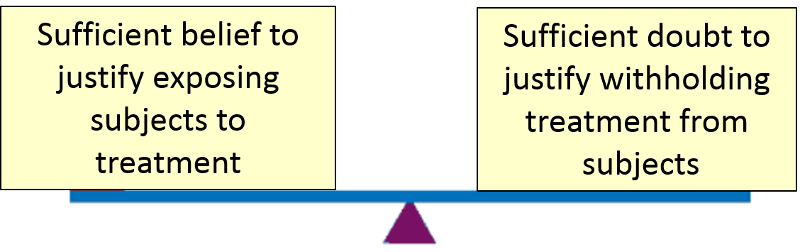
It is unethical to conduct a clinical trial in the absence of equipoise, and if equipoise ceases to exist during the course of a clinical trial, the trial must be discontinued.
Before research on living humans is conducted, a detailed protocol must be submitted to an Institutional Review Board (IRB) for review and approval. This is true not only of clinical trials, but also all other types of human research including case-series, cross-sectional surveys, prospective and retrospective cohort studies, and case-control studies. [For a more detailed overview of the ethical considerations for human research, see our online module on Research Ethics.]
|
"Human Research" is defined as any systematic investigation involving living humans (including research development, testing and evaluation), designed to develop or contribute to generalizable knowledge.
|
Informed Consent
One of the key things that an IRB will consider is whether potential subjects have provided informed consent, which is the process by which study participants consent to be subjects only after becoming fully informed and understand all aspects of the research including the purpose, risks, type of information to be collected, potential benefits, and alternatives to the research. Informed consent should allow people to make a fully informed decision about whether to participate in a study or not based on their own goals and values. Informed consent must be obtained before assignment to a treatment group, and consent can be withdrawn at any time during the study.
Potential participants must be fully informed about:
- The purpose of the study
- The treatment options (including alternatives) and the potential outcomes
- The risks and potential benefit of the study
- Randomization, i.e., that the treatment they receive is not their choice
- What will be required of them (questionnaires, visits, samples collected, etc.)
- Their ability to withdraw from the study at any time without consequence