

Theories on Aging
Why do we age? Although the question has been raised since hundreds of years ago, the mysteries that control human life span are yet to be discovered. During the last decades, many theories have been proposed to explain the process of aging, but neither of them seems to be fully satisfactory. Instead, these theories interact with each other and all of these together contribute to aging. The image below shows different mechanisms that have been proposed to explain aging. In this module, however, we will only discuss three major theories of aging: cellular senescence, DNA damage, and telomere shortening.
Cellular Senescence
Recent studies suggest that cellular senescence might be a cellular model of organismal aging. Accumulations of senescent cells were found in vivo in mammals with increasing age, and at sites of age-related pathology. [Lombard DB, Chua KF et al. :DNA Repair, Genome Stability, and Aging. Cell 2005;120 (4): 497-512.]
Cellular senescence is a state of irreversible growth arrest, which was first introduced by Hayflick and Moorhead in 1961. They found that normal human fibroblasts had a limited replicative potential and eventually entered a state of irreversible growth arrest, meaning that cells being cultured in vitro had lost the ability to divide, despite having ample space and nutrients in the culture medium. Two general models have been proposed to explain how cellular senescence may contribute to aging. First, senescent cells in tissues may accumulate to the point where the strength and functional capacity of tissues in compromised. A second model proposes that senescence in stem cells limit their regenerative potential which eventually leads to a progressive loss of tissue strength and functional capacity. Cellular senescence can be triggered by a number of mechanisms, such as telomere shortening and DNA damage an described below.
limit their regenerative potential which eventually leads to a progressive loss of tissue strength and functional capacity. Cellular senescence can be triggered by a number of mechanisms, such as telomere shortening and DNA damage an described below.
DNA Damage
 Studies showed that cellular senescence is commonly triggered by various forms of DNA damage. Mutant mice that are deficient in DNA repair show premature senescence and progeroid phenotypes, suggesting the involvement of DNA damage-induced senescence in aging. [Collado M, Blasco MA et al.:Cellular Senescence in Cancer and Aging. Cell 2007;130(2):223-233.]
Studies showed that cellular senescence is commonly triggered by various forms of DNA damage. Mutant mice that are deficient in DNA repair show premature senescence and progeroid phenotypes, suggesting the involvement of DNA damage-induced senescence in aging. [Collado M, Blasco MA et al.:Cellular Senescence in Cancer and Aging. Cell 2007;130(2):223-233.]
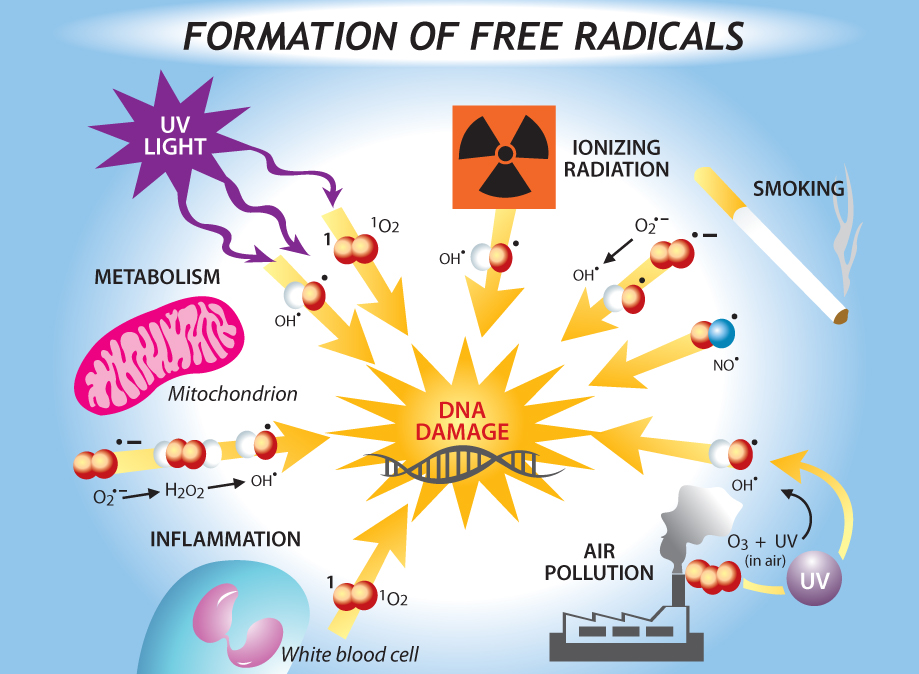
Sources of DNA damage include external sources, such as ionizing radiation tobacco smoke, air pollution and genotoxic drugs, and cell-intrinsic sources, such as replication errors, programmed double-strand breaks, and DNA damaging agents—reactive oxygen species (ROS). The image on the right shows a variety of sources of DNA damage. [Image source: http://churchandstate.org.uk/2013/02/the-first-person-to-live-to-150-has-already-been-born/]
Reactive oxygen species (ROS),such as superoxide anion, hydroxyl radical, hydrogen peroxide and nitric oxide, are normal byproducts of metabolism took place in mitochondria, and are believed to be one important source of DNA damage.
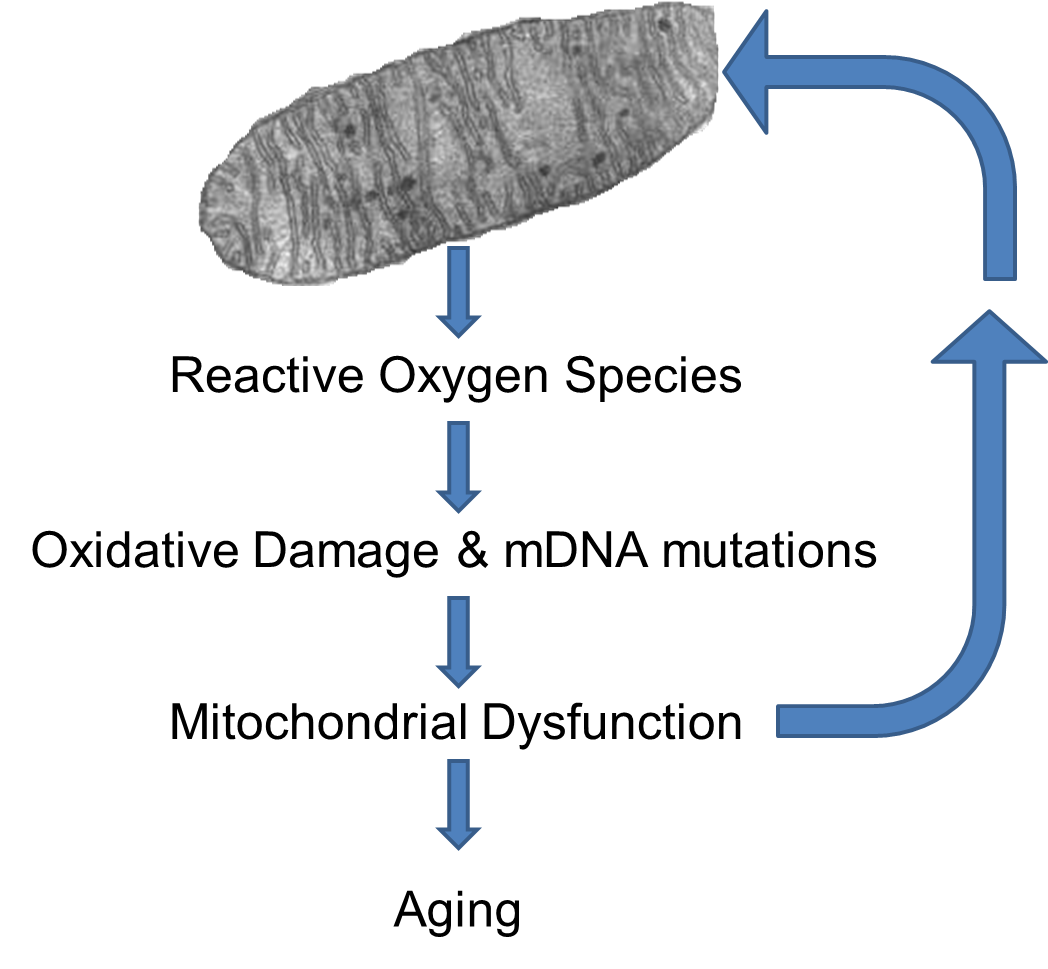
ROS can damage the mitochondria's DNA (mtDNA) and proteins, and the mutant mtDNA in turn are more liable to produce ROS byproducts. Therefore a positive feedback loop of ROS is established. With age the number of mutant mtDNA increase and the mitochondrial functions decline, leading to an increased production of ROS. [Lagouge M, Larsson NG: The Role of Mitochondrial DNA Mutations and Free Radicals in Disease and Ageing. J. Intern. Med. 2013;273(6):529-543.]
The increased generation of ROS can cause lipid peroxidation, protein damage, and several types of DNA lesions in cells. Therefore, ROS are considered important factors in the mechanisms of such diseases as diabetes, cancer, atherosclerosis, heart attacks, Alzheimer's disease, as well as in aging. Evidence has shown that species that live longer generally show higher cellular oxidative stress resistance and lower levels of mitochondrial ROS production compared to species that live shorter.
Telomere Shortening
Telomeres are repeated nucleotides sequences on the end of chromosomes that are believed to protect the DNA strands and prevent them from fusing with other strands. Telomeres lose a little bit of their length during each cell division. Since replicative DNA polymerases are not able to replicate telomeres, and telomerase (specialized DNA polymerase that could replicate telomeres) are not expressed in normal mammalian somatic cells, telomeres become too short to replicate after a fixed number of cell divisions. Eventually, the cell will stop growing and enter cellular senescence. The image below illustrates the telomere shortening as cells divide.
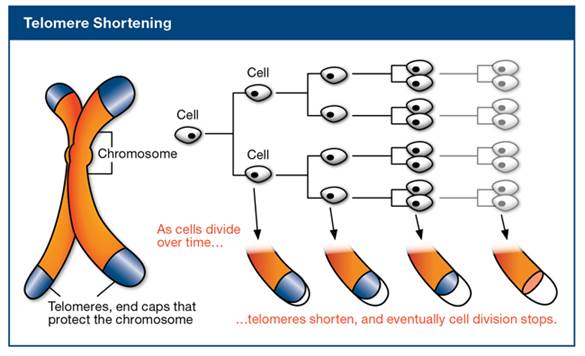
[ Image source: http://www.wholehealthinsider.com/newsletter/2012/a-genetic-solution-to-slowing-aging-and-preventing-disease/]
Telomeres are particularly susceptible to accumulation of DNA damage with age, and this damage at telomeres is restricted to be repaired because of shelterin, a multiprotein complex binding to the telomeres. Shelterin prevent the access of DNA repair proteins to the telomeres. Therefore, DNA damage at telomeres is persistent, inducing cellular senescence efficiently. [López-Otín C, Blasco MA et al.:The hallmarks of aging. Cell 2013;153 (6):1194-1217.]
The video below is a 3D animation on telomeres and telomerase (0:59min).
![]()
![]()
Calorie Restriction
Some studies have shown that calorie restriction (i.e., a 20-40% reduction of dietary caloric intake) extends life expectancy in several species ranging from yeast to rodents. One possible explanation is that caloric restriction reduced production of reactive oxygen species by mitochondria. In addition, calorie restriction induces autophagy that removes harmful proteins and organelles, thereby reducing the accumulating damage to the cell. However, this phenomenon has not yet been demonstrated in humans. For an excellent review, see Ivanova DG, Yankova TM: The free radical theory of aging in search of a strategy for increasing life span. Folia Med 2013;55(1):33-41.