


Prions
The term "prion" was coined from "protein infection particle." Prion diseases have been with us for some time, but only came to the attention of the general public during the epidemic of "Mad Cow Disease" that began in the United Kingdom in the 1970s. Prion proteins are normally found in all mammalian brains, but it is believed that altered forms of these proteins fold abnormally as a result of a mutations; as a result the proteins encoded by the mutant genes have distorted shapes, and they are not broken down normally.
Prusiner's Hypothesis:
(Dr. Stanley Prusiner was awarded a Nobel Prize in 1997 for his work on the discovery of prions.)
His hypothesis is that a mutant prion is an abnormally folded prion protein (PrPSc) that is resistant to heat & sterilization, and does not evoke an immune response. The protein is chemically the same, but folds differently. The abnormal protein contacts normal proteins in neural tissue and induces them to refold into an abnormal conformation as well. Refolded molecules induce the same change in still more proteins. The abnormal proteins resist degradation and accumulate in neural tissue causing damage. The illustration below shows an artists depiction of a normal prion protein (PrPC on the left) and an abnormal prion protein (PrPSc on the right). The abnormally folded protein has segments folded into "beta sheets" (the segments shown with thick green arrows). These segments tend to sticky together causing clumps of proteins that resist breakdown. Over time, the clumps grow larger and destroy nerve cells in areas of accumulation, leading to progressive neurological symptoms (so-called "prion diseases").
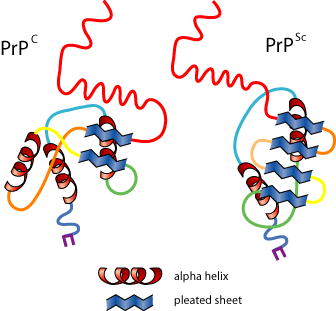
Source: http://www.scq.ubc.ca/prions-infectious-proteins-repsonsible-for-mad-cow-disease/
Human Prion Diseases
Animal Prion Diseases
- Scrapie
- Bovine Spongiform Encephalopathy (BSE)
- Feline spongiform encephalopathy
The deposition and accumulation of these abnormal proteins in the brain results in brain tissue that appears to be riddled with holes when the brain is sectioned and stained post-mortem. As a result, these disease are also referred to as transmisible "spongiform " encephalopathies. The image below shows a specimen of brain tissue viewed through a microscope. The light colored "holes" represent areas of the brain where deposits of the abnormal proteins destroyed cells.
" encephalopathies. The image below shows a specimen of brain tissue viewed through a microscope. The light colored "holes" represent areas of the brain where deposits of the abnormal proteins destroyed cells.
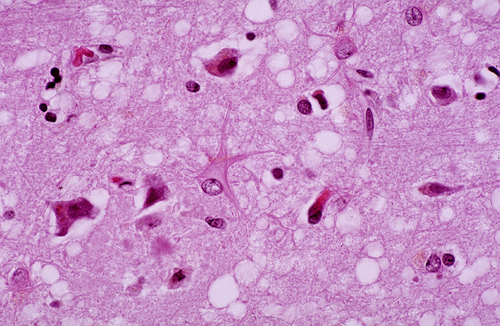
Source: http://neuropathology-web.org/chapter5/chapter5ePrions.html
The abnormal folding is believed to be initiated by a mutation. In the case of Creutzfeldt-Jakob Disease (CJD) there are two forms of the disease:
- Sporadic CJD, which occur very rarely as a result of random mutations in the brain; this form is not passed on to family member.
- Familial CJD, which occurs as a result of a mutation in the cells giving rise to sperm or ova; this form can be passed on from generation to generation.
In addition, prion diseases are also transmissible if neural tissue with abnormal prion proteins is ingested or transmitted through medical procedures such as transplants or injections or via instruments contaminated with abnormal prion proteins. Kuru is a type of transmissible spongiform encephalopathy that occurred in New Guinea as a result of ritual cannibalistic practices that involved eating the brain tissue of human relatives who had died. At some point a mutation resulted in an abnormal prion protein that was transmitted among relatives as a result of eating affected brain tissue, which initiated a chain reaction in the the recipients that resulted in transmissible spongiform encephalopathy.
Scrapie is a disease of sheep that was described some time ago, and is now thought to be a prion disease. Many believe that the epidemic of bovine spongiform encephalopathy (BSE or "mad cow disease") that occurred in the United Kingdom in the 1970s was triggered when farmers began to feed cattle meat and bone meal that contained neural (nerve) tissue from scrapie-infected sheep or from cattle with spontaneously occurring BSE.
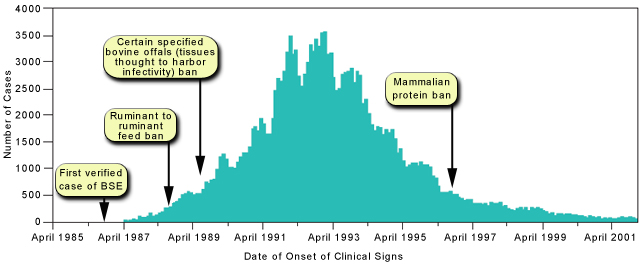
Source: http://neuropathology-web.org/chapter5/chapter5ePrions.html
The epidemic in cattle peaked in 1993 and is believe to have caused more than 184,500 cases of BSE. In conjunction with that epidemic a new human prion disease called variant Creutzfeldt-Jakob disease (vCJD) was first reported by the United Kingdom in 1996. There is evidence that vCJD resulted when humans ate meat or other animal tissues from cattle infected with BSE.
Link to CDC's web pages on prion diseases.