The Biology of Cancer
Contributors:
The oldest descriptions of cancer were written in Egypt as early as 3000 B.C., as part of an ancient Egyptian textbook on surgery. The name, "cancer" comes from the Greek word carcinos, which means crab. Hippocrates used this term to describe the disease because of the projections of a cancer invading nearby tissues. During the 16th century, when the theory of bodily humors prevailed, it was believed that an excess of black bile caused cancer. The renowned anatomist Andreas Vesalius searched diligently for this black bile and ultimately discarded the this theory when he was unable to find it. In 1838 a botanist named Matthias Schleiden and Theodor Schwann, a physiologist, proposed that all living things were composed of fundamental units called cells. Shortly after the introduction of this idea, Virchow (the "father" of pathology) proposed that cells only arose from other cells and that growth could only occur as a result of hypertrophy or hyperplasia. Virchow studied cancers under with a microscope and recognized that they represented hyperplasia in an extreme form that he dubbed "neoplasia."
Evidence accumulated to support the idea that cancer was the result of uncontrolled cell division, but the cause was unknown. The earliest clues came from epidemiologic observations. For example, in 1775 Dr. Percival Pott, an English surgeon, reported on the unusual occurrence of scrotal cancer in men who had worker as chimney sweeps as boys, and he speculated that the soot, tars, and dirt to which the chimney sweeps were chronically exposed may have played a role. Another Englishman, John Hill, warned that excessive use of snuff or chewing tobacco might lead to nasal cancer or cancers of the mouth, tongue, or lip. Nevertheless, it wasn't until the last three decades of the the 20th century that the biological origins of cancers began to be revealed. This module will provide our current understanding of the biological basis of cancer, determinants of the disease, and ways to prevent or reduce the risk of getting cancer.
After successfully completing this module, you will be able to:
1. Cell differentiation
2. Benign tumor
3. Malignant tumor
4. Hyperplasia
5. Dysplasia
6. Metastasis
7. Carcinogen
8. Proto-oncogene and oncogene,
9. Tumor-suppressor gene (anti-oncogene)
10. Apoptosis and suicide gene
1. Genetics
2. Hormones
3. Diet
4. Environmental exposures
5. Ultraviolet light
6. Viral and bacterial infections

Virchow was correct when he concluded that cells arise from others cells, i.e., new cells are born through the division of one cell into two through the process of mitosis. The need for new cells continues throughout our lives, but it is greatest in early life. A fertilized egg divides into two cells, which give rise to four, and those give rise to eight, and then to 16, and 32, and 64, and so on. In a fully grown adult, of course, the rate of cell proliferation is much less, and under normal circumstances, cell division in an adult takes place only when signals indicate the need to replace cells that have been lost, damaged, or worn out.
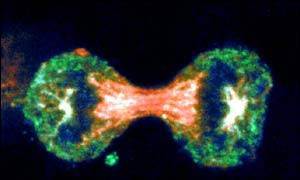
Source: http://news.bbc.co.uk/2/hi/health/4902908.stm
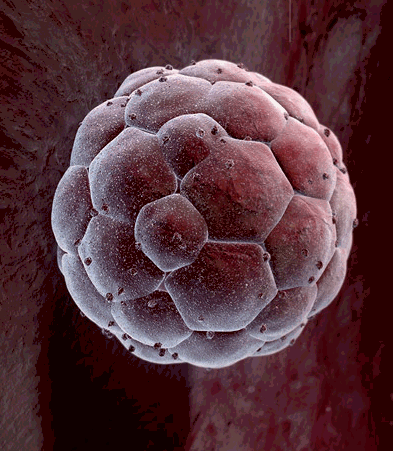
Another big difference between cells in a growing embryo and cells in an adult is that most of an adult's cells are differentiated - they have become specialized in structure and function. Muscle cells are elongated and contain and abundance of contractile proteins, whereas pancreatic cells are specialized for secretion of digestive enzymes or, in the case of pancreatic beta cells, for the synthesis of insulin. In contrast, the cells in the early morula stage of an embryo (shown below to the left) consists of cells that are totipotent - they have the capacity to divide and give rise to any of the specialized cells in the body. In the adult, however, the replacement of shed or worn out cells takes place by division of somatic stem cells (also called adult stem cells), which are not fully differentiated, but can give rise to only a limited array of cells.
|
Source: http://imgarcade.com/1/morula-stage/ |
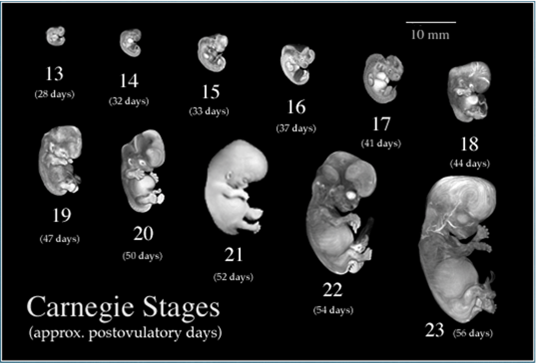
Source: http://embryo.soad.umich.edu/carnStages/carnStages.html |
Source: http://ribbu.com/cool-pictures/human-anatomy/ |
For example, hematopoetic stem cells in the bone marrow can divide and give rise to progenitor cells that can differentiate into cellular element of blood and the immune system, including red blood cells, lymphocytes, neutrophils, eosinophils, basophils, monocytes, and platelets. Bone marrow stromal stem cells (also called mesenchymal stem cells, or skeletal stem cells) can generate bone, cartilage, and fat cells. Whenever stem cells are called upon to generate a particular type of cell, they undergo an asymmetric cell division in which one of the daughter cells has a finite capacity for cell division and begins to differentiate, whereas the other daughter cell remains a stem cell with unlimited proliferative ability.
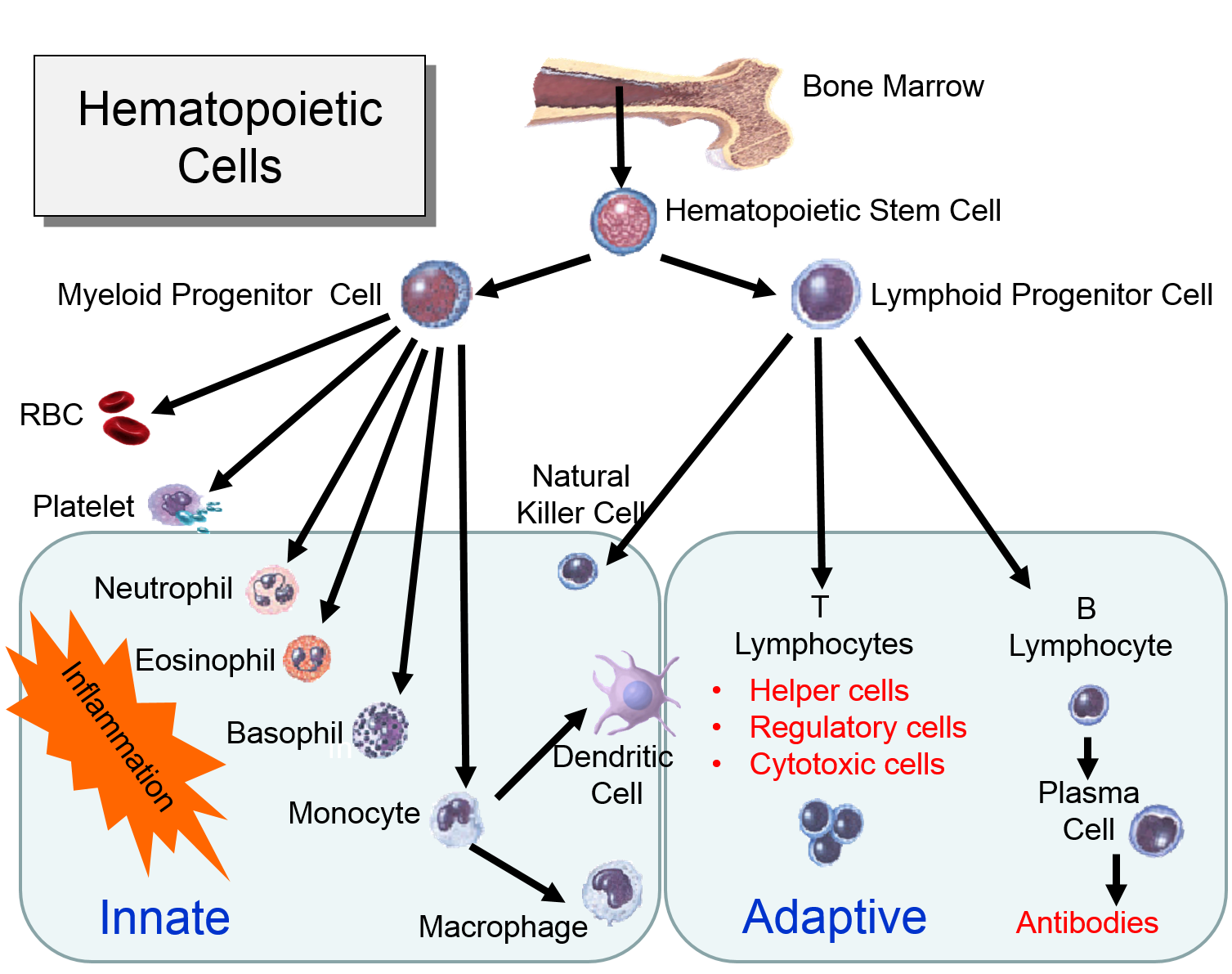
Watch the video below to see a short (2 min 18 sec) explanation of hematopoetic stem cells and somatic stem cells in the intestine.
Virchow was correct when he concluded that cells arise from others cells, i.e., new cells are born through the division of one cell into two through the process of mitosis. The need for new cells continues throughout our lives, but it is greatest in early life. A fertilized egg divides into two cells, which give rise to four, and those give rise to eight, and then to 16, and 32, and 64, and so on. In a fully grown adult, of course, the rate of cell proliferation is much less, and under normal circumstances, cell division in an adult takes place only when signals indicate the need to grow or to replace cells that have been lost, damaged, or worn out. In addition, most cells in an adult will be differentiated to serve a particular purpose. These processes - cell division and differentiation - are tightly regulated by many signals and signal pathways.
Cell differentiation is incompletely understood, but it involves the activation or inactivation of certain genes in response to the cell's interactions with its neighboring cells and with its extracellular matrix (ECM). For example, receptors on the cell will bind to specific molecular elements in the ECM, and this binding activates intracellular signal transduction pathways that turn certain genes on or off. As a result of these interactions, some genes can be expressed in a given cell, but others cannot. For example, in a muscle cell, the genes that encode the contractile proteins actin and myosin are activated, but the gene encoding for insulin synthesis is inactivated. Some cells, e.g., skeletal muscle cells and nerve cells, become terminally differentiated, meaning that their ability to proliferate is permanently lost, although they continue to perform their specialized functions for a long time. Terminally differentiated cells like these (i.e., mature muscle or nerve cells) do not form tumors, although less differentiated myoblastic or neuronal stem cells may form tumors. Cell-cell and cell-ECM interactions are important not only for the induction of differentiation, but also for maintenance of differentiation in some cell types. One of the hallmarks of tumor cells is that they lose their ability to sense the ECM or neighboring cells.
The diagram to the right summarizes events leading to cell division.
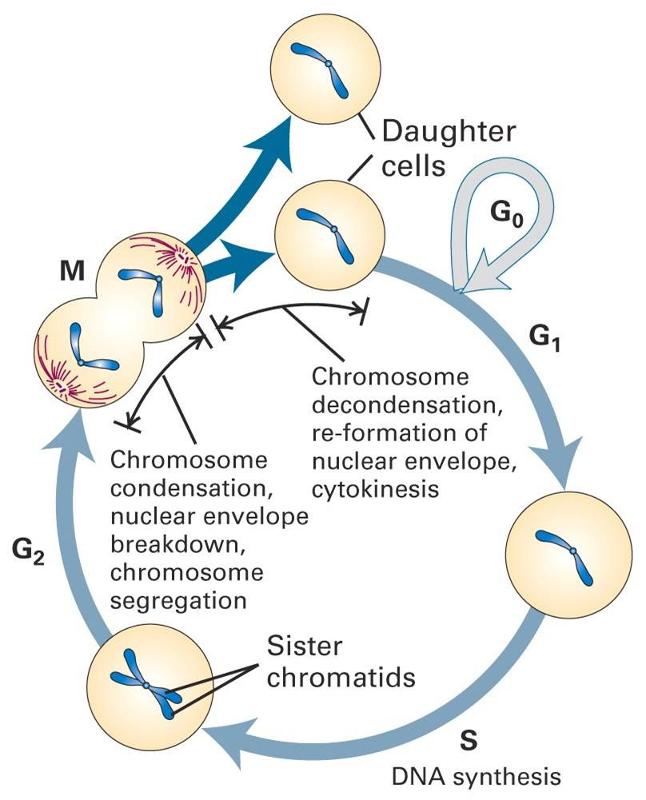
Many cells in an adult are not actively in the process of replicating; this is depicted in the diagram as "cells that cease division," also known as the G0 phase or the "resting phase." The term "resting phase" is a misnomer, however, because the cell is actively carrying out its normal specialized functions, and It is only resting in the sense that it isn't actively dividing. If conditions require additional cells, the cell will receive signals that promote cell division. These signals will push the cell to complete the G1 phase (cell enlargement) and proceed to the S-phase, during which DNA is replicated. In the G2 phase the cell prepares for division by increasing in size and replicating intracellular organelles. It then divides through mitosis (the M-phase). In a sense, the critical juncture is the transition from G1 to the S-phase. This transition is carefully regulated by multiple factors, some of which promote the transition. Genes known as proto-oncogenes can be switched on to produce proteins that protein the transition to the S-phase. Counteracting this push to reproduce are genes known as anti-oncogenes (also called tumor suppressor genes) that inhibit transition to the S-phase. The video below provides a short visual summary of these events, known as the cell cycle.
The two videos below summarize the signaling events that regulate the cell cycle and events occurring during the cell cycle.
For more detail see Michael Andreeff, MD, PhD, David W Goodrich, MD, and Arthur B Pardee, MD. Chapter 2 - "Cell Proliferation, Differentiation, and Apoptosis" in Cancer Medicine, 6th Edition"
Apoptosis is also referred to as programmed cell death. It is an essential process for removing cells that are stressed, damaged, or worn out. It is estimated that over 50 billions cells undergo apoptosis each day in adults. Apoptosis is also carefully regulated through complex mechanisms. Mutations that affect these regulatory pathways have the potential to contribute to carcinogenesis by failing to eliminate abnormal neoplastic cells or by failing to eliminate cells with other mutations that are premalignant. Defects in apoptosis can also confer resistance to chemotherapy, radiation, and immune-mediated cell destruction.
Proto-oncogenes, anti-oncogenes (tumor suppressor genes), and apoptosis, play a central role in understanding the pathogenesis of cancer. In short, mutations and inherited abnormalities can cause these regulatory control mechanisms to become dysfunctional. As you will see, mutations in any of these mechanisms can cause a cell to divide or survive longer than normal, and if multiple mutations affecting these regulatory mechanisms accumulate in a single cell, the cell will have lost all control with respect to cell division. This single cell, dividing repeatedly and without regulation will create an ever expanding clone of cells which will also undergo unregulated cell division. This is the essence of cancer.
The outer layer of skin (epidermis) is about 12 cells thick. Cells in the basal layer (bottom row) divide just fast enough to replenish cells that are shed. When a basal cell divides, it produces two cells. One remains in the basal layer and retains the capacity to divide. The other migrates out of the basal layer and loses the capacity to divide. The number of dividing cells in the basal layer, therefore, stays about the same.
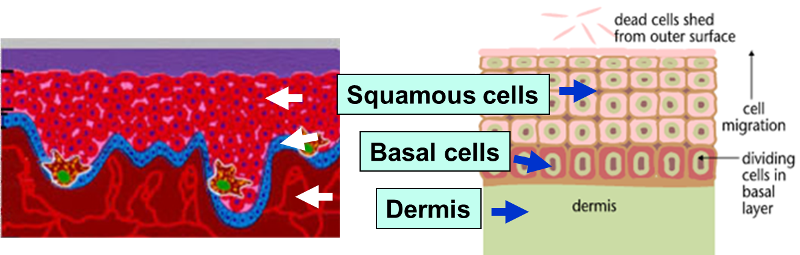
The transition to skin cancer begins when the normal balance between cell division/cell loss is disrupted. Basal cells divide faster than needed to replenish the cells being shed, and with each division both of the two newly formed cells will often retain the capacity to divide, leading to an increased number of dividing cells.
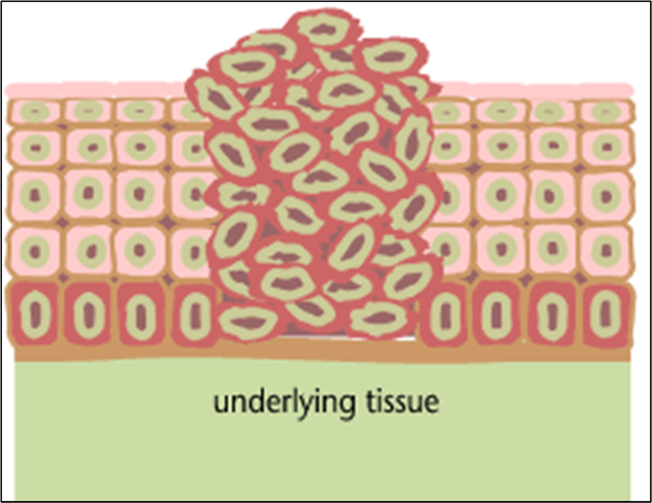
This creates a growing mass of tissue called a "tumor" or "neoplasm." As more and more dividing cells accumulate, the normal organization of the tissue gradually becomes disrupted.
Benign tumors (e.g. skin moles, lipomas): abnormal growths that are no longer under normal regulation, but they grow slowly, resemble normal cells, and still have surface recognition proteins that bind them together and keep them from invading or metastasizing.
Source: http://www.playbuzz.com/roberts32/what-is-the-cellular-basis-of-cancer
1775
The initial clues about the pathogenesis of cancer came from both epidemiologic and laboratory observations. In 1775 Percival Pott, a surgeon at St. Bartholomew's Hospital noticed a marked increase in cases of scrotal cancer in his clinic. His patients were almost invariably chimney sweeps or "climbing boys" - poor indentured orphans who apprenticed as sweeps and were sent up chimneys to clean the flues of ash, often naked and covered in oil. He noted that they spent hours in contact with grime and ash and they had particles of soot embedded in their skin. In 1788 the Chimney Sweepers Act was passed in Parliament, preventing master sweeps from employing children under the age of 8. In 1834 the age was raised to 14, and in 1840 it was raised to 16. By 1865 the use of young climbing boys was forbidden. This simple observation was important, because it suggested that an environmental exposure could play a role in causing cancer.
1902
German scientist Theodor Boveri proposed that cancer could develop from a single cell with a damaged chromosome.
1911
Peyton Rous demonstrated injection of an extract containing a virus could induce a cancer, specifically a sarcoma, in live chickens.
1920s
In the 1920s, several US watch companies produced "glow in the dark" dials painted with radioluminescent paint that contained radioactive radium. The dials were painted by hand with small brushes, and in order to maintain a precise shape to the brush, the employees "pointed" the brushes on their tongues. The radium, ingested in small doses over time accumulated in bone and produced bone cancer in many of the employees (mostly young women).
1950
As time went on there were similar observations suggesting a link between environmental agents and cancer. In 1950 Wynders and Graham conducted one of the earliest case-control studies suggesting a link between tobacco smoking and lung cancer. A similar conclusion was reached by Richard Doll and Bradford Hill. They argued that a chemical in tobacco smoke caused lung cancer, but they were unable to explain the mechanism.
|
From "The Emperor of All Maladies" by Siddhartha Mukherjee: "By the early 1950s, cancer researchers had thus split into three feuding camps. The virologists, led by Rous, claimed that viruses caused cancer, although no such virus had been found in human studies. Epidemiologists, such as Doll and Hill, argued that exogenous chemical caused cancer, although they could not offer a mechanistic explanation for their theory or results. The third camp, of Theodor Boveri's successors, stood at the farthest periphery. They possessed weak, circumstantial evidence that genes internal to the cell might cause cancer, but had neither the powerful human data of the epidemiologists nor the exquisite experimental insights of the chicken virologists. Great science emerges out of great contradiction, and here was a gaping rift slicing its way through the center of cancer biology. Was human cancer caused by an infectious agent? Was it caused by an exogenous chemical? Was it caused by an internal gene? How could the three groups of scientists have examined the same elephant and returned with such radically variant opinions about its essential anatomy?" "But the set of disparate observations - from Blumberg to Ames to Warren and Marshall - could not simply be stitched together into a coherent theory of carcinogenesis. How could DES, asbestos, radiation, hepatitis virus, and a stomach bacterium all converge on the same pathological state, although in different populations and in different organs?" |
1959
A virologist named Howard Temin added Rous sarcoma (RSV) virus to cells in culture and found that the cells began to divide uncontrollably. In addition, the viral genome had inserted itself into the DNA of the cells. This was particularly remarkable, since the genome of Rous sarcoma virus is contained in single-stranded RNA. Temin postulated that Rous sarcoma virus was able to convert its single-stranded RNA into a double-stranded DNA form that could be inserted into the DNA of the infected cells. Several years later Temin and Satoshi Mizutani isolated an enzyme, which is now called reverse transcriptase, which is able to create this transformation. Reverse transcriptase is the enzyme which HIV uses to insert itself into the genome of infected lymphocytes in humans.
In the aftermath of these findings many cancer biologists searched for evidence of retroviruses in human cancers, but none were found. Temin reasoned that the Rous sarcoma virus had caused cells to become cancerous by causing genetic alterations in infected cells. However, it was possible that the genetic alterations weren't necessarily caused just by a virus.
|
Src: A Protein Kinase In the 1970s virologists began making mutants of RSV, which is a small virus with only 4 genes. Some of the mutants were able to replicate, but were unable to cause cancer, and with these experiments, they were able to identify which gene in RSV was responsible for causing cancer. The gene was called"Src", short for sarcoma. And because it cause a cancer, it was dubbed an "oncogene." Later studies revealed that Src encoded for a protein kinase. Protein kinases are a family of proteins that act as on-off switches by attaching a phosphate group to particularly enzymes. Attaching a phosphate to one enzyme might activate it, for example. Often a kinase activated another kinase, which in turn tagged another kinase with phosphate and activated it, and this in turn might activate another in a chain reaction such that the switching on became amplified in a powerful way. This sequence of events might then reconfigure a cell from a non-dividing state to a dividing state. Mutant Src produced an abnormally hyperactive kinase that phosphorylated all kinds of kinases within the cell and therefore turned on many of the molecular switches, including those that controlled cell division. Normal cellular Src phosphorylated the same kinases, but at a slower, more normal rate that was carefully regulated. These observations suggested the possibility that this cancer causing viral Src was actually a normal cellular gene with had mutated. As a result, one could think of the normal precursor gene as the "proto-oncogene," which at some pointed mutated to give rise to the Src oncogene. Scientists subsequently found that a family of similar genes was present in virtually all cells.
|
1959
In the late 1950s Peter Nowell and David Hungerford found a small abnormality in the chromosomes of patients with chronic myelogenous leukemia (CML). At the time techniques for imaging chromosomes were still crude, but it appeared that a small portion of one copy of chromosome #22 was missing. They dubbed this the"Philadelphia" chromosome, since that is where they discovered it. This provided further weight to the idea that genetic alterations (mutations) could be involved in the occurrence of cancers.
1963
A pathologist, Oscar Auerbach, published a study based on 1522 autopsies of smokers and non-smokers. A careful examination of the cells lining the airways of smokers showed a spectrum of pathological changes ranging from swelling and thickening, to cells with premalignant changes (atypia or dysplasia), to clusters of cells that were malignant and represented invasive carcinoma. This array of changes suggested to Auerbach that cancer evolved through a progression of changes from normal to cancerous over a long period of time.
Late 1960s
Bruce Ames, a bacteriologist at Berkeley, was studying mutations in Salmonella and observed that mutations could enable or disable the growth of bacteria on a petri dish. A strain of Salmonella normally unable to grow on galactose could acquire a gene mutation that enabled it to do so. By counting the number of growth enabled colonies Ames could quantify the mutation rate. Bacteria could be exposed to a certain chemical and Ames could then measure the mutation rate. He also noticed that chemicals that were mutagens also tended to be carcinogens. Dye derivatives that were known to be potent human carcinogens caused hundreds of mutations in the bacteria, as did x-rays, benzene compounds, and nitrosoguanidine, all of which caused cancers in rats and mice. Some known carcinogens did not induce mutations in Ames's in vitro system, but his findings did provide evidence of a link between mutations and cancer.
Late 1960s
Baruch Blumberg, a biologist, found evidence that viruses caused cancer. Patients with chronic hepatitis B infection had a 5-10 fold increase in risk of developing liver cancer. Blumberg concluded, "The inflammation induced by the virus in liver cells, and the associated cycle of death and repair, appeared to be responsible for the cancer...."
1971
Herbst et al. published results of a case-control study indicating that the drug diethylstilbesterol caused vaginal cancer in females who had been exposed to the drug in utero. .
1971
Alfred Knudson conducts an analysis that provides evidence that a mutation is responsible for retinoblastoma, a cancer of the eye that can occur in children. Retinoblastomas follows were known to occur in two distinct patterns: a familial form, which is seen with high frequency in some family lines, and a sporadic form. The inherited form occurs early and is typically diagnosed within the first 2-6 months after birth, while the sporadic form typically occurred 2-4 years after birth. Since humans have two copies of each gene, Knudson hypothesized that individuals with the familial form inherited one defective allele, which by itself was not enough to cause the cancer. With one inherited defect, however, he proposed that a mutation in the other allele could trigger the familial form of retinoblastoma. Individuals without an inherited defect would require two mutations, one in each of the two alleles in order to produce the sporadic form of the cancer. As a result, individuals with the sporadic form tended to develop retinoblastoma later in life. Knudson called this the two-hit hypothesis of cancer. The mutated gene that was responsible was dubbed "Rb."
|
Key Concept: Proto-Oncogenes and Anti-Oncogenes
The retinoblastoma gene (Rb) is an anti-oncogene (or tumor suppressor gene); it produces a protein whose normally function is to bind to several other proteins which prevents them from activating cell division. When a cell receives normal signals to divide, it inactivates Rb by tagging it with a phosphate group. Anti-oncogenes act like recessive genes; if one of the two genes is rendered non-functional by a mutation, there is no change in cell behavior, because the other gene is still functioning. However, if both copies of the tumor suppressor gene are knocked out, the "brakes" to cell proliferation no longer function. In the case of Rb, some persons are born with one defective copy, so they are predisposed. If the other copy mutates in a somatic cell, then they can develop a tumor.
In contrast, Src is a proto-oncogene, which normally serves to activate cell division when the cell receives an appropriate signal, whereas the mutant form of the gene (an oncogene) causes unrestrained activation.
Michael Bishop and Harold Varmus, demonstrated that precursors of oncogenes - proto-oncogenes - existed in all normal cells. Harold Varmus, Michael Bishop, and Alfred Knudson subsequently proposed that these two abnormalities, activated proto-oncogenes and inactivated tumor suppressors, represented the critical defects in a cancer cell. |
1973
Scientist Janet Rowley determines that the defect in patients with chronic myelogenous leukemia ("the Philadelphia chromosome") is actually a translocation in which the head of chromosome 22 is fused to the tail of chromosome 9 to create a novel gene.
1982
Following up on the discovery of the Philadelphia chromosome, a team of Dutch researchers isolated the gene on chromosome 9 and called it "abl". Two years later the gene on chromosome 22 was isolated and called "Bcr". The oncogene created by their fusion was called Bcr-abl. In 1987 David Baltimore's lab in Boston created a transgenic mice with the fused gene, and they developed fatal leukemia. Follow up studies then determined that Bcr-abl encoded for a protein kinase (a protein that regulated other proteins by tagging them with a phosphate group.
1982
Robert Weinberg, a virologist working at MIT, and Chiaho Shih, a graduate student in his lab, used gene transfer techniques to insert fragments of DNA from human bladder cancer cells into normal cells and identified clusters of cells that had begun to proliferate abnormally. Weinberg and two other labs, working independently published their findings regarding a cancer causing gene, called "ras" from human cancer cells.
|
From "The Emperor of All Maladies" by Siddhartha Mukherjee: "Like src, ras was also a gene present in all cells. But like src again, the ras gene in normal cells was functionally different from the ras present in cancer cells. In normal cells, the ras gene encoded a tightly regulated protein that turned 'on' and 'off' like a carefully modulated switch. In cancer cells, the gene was mutated, just as Varmus and Bishop had predicted. Mutated ras encoded a berserk, perpetually hyperactive protein permanently locked 'on'. This mutant protein produced an unquenchable signal for a cell to divide - and to keep dividing. It was the long-sought 'native' human oncogene...." |
1983-1993
A number of other oncogenes and tumor suppressor genes were found to be involved in human cancers.
1984
Transgenic technology allowed scientists to introduce exogenous genes into early mouse embryos to create "transgenic mice" in which one or more genes were artificially and permanently modified. When Philip Leder's lab introduced the oncogene c-myc into mouse breast cells, it produced only small tumors, and typically the tumors only occurred after pregnancy, suggesting that hormonal influences played a role. Leder then created a transgenic line with two oncogenes: ras and myc. These grew multiple tumors, but Leder was puzzled because millions of breast cells had acquired ras and myc, but only a few dozen formed tumors.
1988
Burt Auerbach and others had demonstrated the lung cancer, cervical cancer, and colon cancer evolved slowly over time progressing from hyperplasia to dysplasia to carcinoma in situ, and eventually to invasive carcinoma. Bert Vogelstein at Johns Hopkins Medical School collected specimens from patients who had different stages of colon cancer and tested them for the presence of four genes that coded for oncogenes or tumor suppressors. He found that the stages of cancer progression correlated with the activation of oncogenes and the inactivation of tumor suppressor genes.
|
Proto-oncogenes and tumor suppressor genes encode for proteins that ultimately have effects on the cell cycle. However, it is well known that regulatory proteins often have their effect by modifying another protein, which in turn modifies yet another protein, and so forth. This cascade of events is referred to as a signaling pathway. "Proto-oncogenes and tumor suppressor genes, cancer biologists discovered, sit at the hubs of such signaling pathways. Ras, for instance, activates a protein called Mek. Mek in tern activates Erk, which through several intermediary steps, ultimately accelerates cell division. This cascade of steps, called the Ras-Mek-Erk pathway - is tightly regulated in normal cells, thereby ensuring tightly regulated cell division. In cancer cells, activated "Ras" chronically and permanently actives Mek, which permanently activates Erk, resulting in uncontrolled cell division - pathological mitosis." [Source: "The Emperor of All Maladies" by Siddhartha Mukherjee]
But the activated ras pathway (Ras-Mek-Erk) does not merely cause accelerated cell division; the pathway also intersects with other pathways to enable several other "behaviors" of cancer cells. |
1990s
At Children's Hospital in Boston in the 1990s, the surgeon-scientist Judah Folkman demonstrated that certain activated signaling pathways within cancer cells, ras among them, could also induce neighboring blood vessels to grow. A tumor could thus "acquire" its own blood supply by insidiously inciting a network of blood vessels around itself and then growing, in grape like clusters, around those blood vessels, a phenomenon that Folkman called tumor angiogenesis. Folkman's Harvard colleague Stan Korsmeyer found other activated pathways in cancer cells, originating in mutated genes, that also blocked cell death, thus imbuing cancer cells with the capacity to resist signals. Other pathways allowed cancer cells to acquire motility, the capacity to move from one tissue to another - initiating metastasis. Yet other gene cascades increased cell survival in hostile environments, such that cancer cells traveling through the bloodstream could invade other organs and not be rejected or destroyed in environments not designed for survival."
1994
The BRCA2 gene is discovery. It is a tumor suppressor which, if mutated, increase the risk of breast cancer.
1990s
The discovery of the Philadelphia chromosome and the subsequent discovery of Bcr-abl triggered a cascade of events along a signaling pathway with the cell. In normal cells the two separate genes were carefully regulated, but when they were fused, the result was a highly overactive kinase that signaled cells to divide continually. Humans normally make about 500 different kinases which regulate a wide variety of cellular pathways. By tagging specific proteins with a phosphate, they basically switch a pathway on or off. Scientists at the Ciba-Geigy pharmaceutical company in Basel, Switzerland began trying to synthesize drugs that could inhibit these kinases by binding to the protein and blocking its kinase activity. By the early 1990s they had created dozens of them. These compounds were also found to have specificity, meaning that one drug might block the "abl" kinase but not block the "src" kinase. An oncologist at Dana-Farber Cancer Center was able to obtain CGP57148, a kinase blocker that was specific for Bcr-abl. When the drug was added to chronic myelogenous leukemia (CML) cells growing in the lab, the cells died overnight. And the drug (Gleevec) was given to mice that had tumors from implanted CML, the tumors regressed in days, but normal cells were undamaged.
2001
Licensing of Gleevec - Ciba-Geigy merged with Sandoz to form Novartis, but Novartis was reluctant to begin clinical trials for Gleevec, mainly out of concern that the cost of the trials would be prohibitive given the relatively small market for the drug. Eventually, they agreed to make a small amount of the drug and tested an initial group of 54 patients, 53 of whom responded with prompt remissions which were usually long lasting. The drug, now called Gleevec, has become the standard of care for patients with CML. It was licensed in 2001.
|
From The Emperor of All Maladies:
"Gleevec opened a new door for cancer therapeutics. The rational synthesis of a molecule to kill cancer cells - a drug designed to specifically inactivate an oncogene...." |
2005
Unfortunately, some CML patients treated with Gleevec eventually developed cancer cells that were resistant to the drug. These cells had acquired a mutation that altered the structure of the Bcr-abl in such a way that Gleevec would no longer bind to it, although it was still able to switch on cell division. However, in 2005 a second drug was developed that was able to bind to Gleevec-resistant Bcr-abl. The new drug is called dasatinib, and it was able to induce remission in Gleevec-resistant cases. Since then, many more cancer-targeted drugs have been developed. These drugs differ from the original chemotherapeutic drugs in that the newer drugs target cancer cells in a highly specific way that is custom-designed to neutralize the defect in the cancer cells. This highly specific attack means that normal cells are unaffected, resulting in far fewer side effects.
2005
A number of authors recognized that the mortality rates for some of the major forms of lung cancer, including lung, breast, colon, and prostate, had been slowly, but steadily declining over the past 15 years. The reasons for the decline differed among the major cancer types.
Cancer is a clonal disease, meaning that accumulated mutations in a single cell lead to unregulated proliferation, a loss of differentiation, and abnormal behavior. With unregulated proliferation, a single cell will give rise to an ever increasing clone of similarly abnormal cells, although the progeny cells may acquire still more mutations over time. The gene abnormalities (mutations) may be inherited (for example, defective Rb in the familial form of retinoblastoma) or they may be acquired as a result of viral infection or damage to cellular DNA caused by carcinogens, e.g., chemicals in cigarette smoke, asbestos, radiation (UV, radon gas, X-rays). It is also possible for mutations to occur as random errors during cell division. There are cellular mechanisms that repair defects in DNA, but repair is not always successful.
In the short video clip below Dr. David Sherr, from the environmental health department at BUSPH explains what a carcinogen is.
The preceding pages make it clear that cancer evolves as the result of an accumulation of mutations within a single cell. Cells have enzymes whose function is to repair defects in DNA. The genes that encode for these enzyme repair mechanism can also be damaged by mutations. In addition, there are several inherited defects in DNA repair that increase the likelihood of transition to malignancy. For example, xeroderma pigmentosum is an inherited defect in DNA repair that causes extreme sensitivity to ultraviolet light, making them extremely vulnerable to sunburn and skin cancers. Ultraviolet light penetrates the superficial layers of the skin, and causes damage to DNA when the radiant energy is absorbed. For more information on ultraviolet light and adverse effects of overexposure see the module on Sun Exposure.
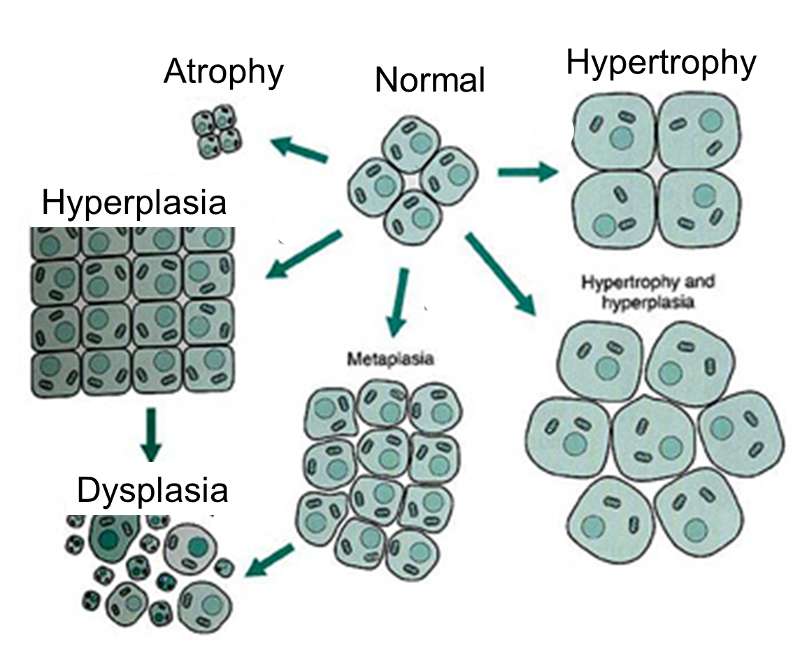
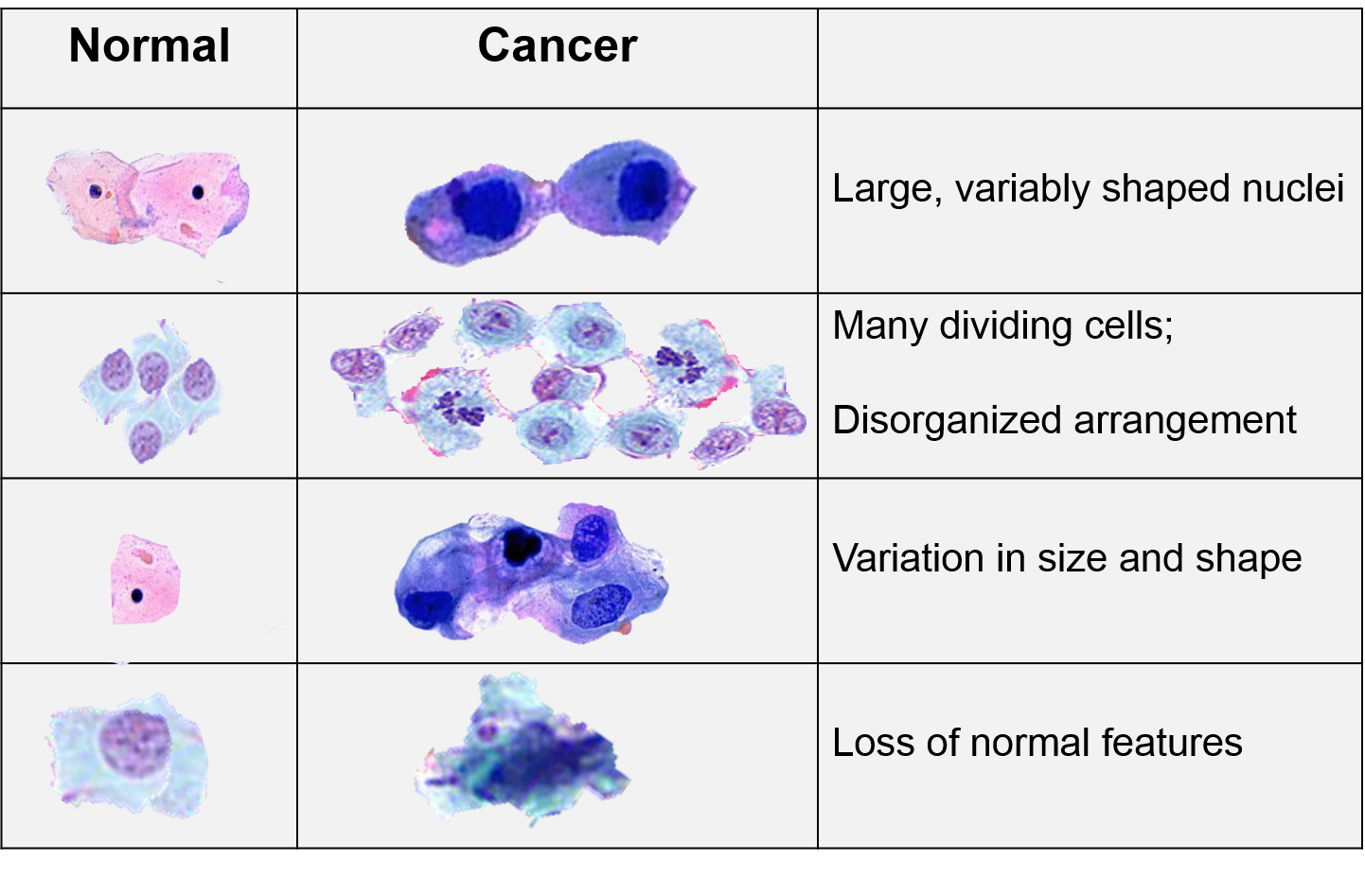
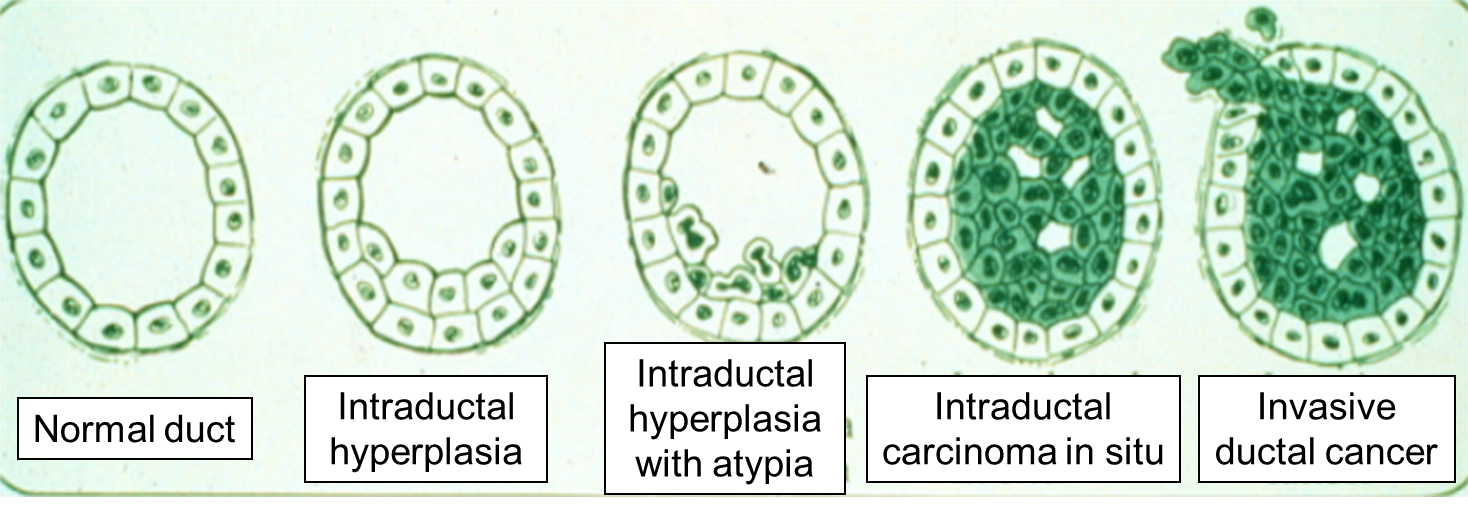
As mutations accumulate in a given cell, there is a progressive loss of regulation of the cell cycle, differentiation, and cell-to-cell adhesion and interaction. These changes are accompanied by progressive abnormalities in morphology (appearance) of the cells. At times, changes in cellular appearance or number are normal responses to physiologic stresses. For example, failure to exercise a muscle will result in atrophy, while repeated exercise produces hypertrophy. Increases in the number of cells can also be temporary responses to stress of some type. For example, pressure on the later aspect of the foot can result in a callus. This would be characterized as hyperplasia, in which there is an increase in the number of cells, but the morphology of the cells and their relationship to one another remains normal. Hyperplasia is a normal response to a specific stimulus, and the cells of a hyperplastic growth remain subject to normal regulatory control mechanisms. On the other hand, dysplasia is a term used by pathologists to describe a spectrum of abnormalities that are indicative of a pre-cancerous state. When dysplasia is present, there is abnormal proliferation, and the cells have a distinctly abnormal and variable appearance; cells vary in shape and size, and there is evidence that cell to cell interaction has broken down resulting in a disorganized appearance of the tissue. The illustration below summarizes these changes in cell morphology.
Dysplasia is illustrated in the histologic section from a biopsy of the uterine cervix shown below. The area to the right encircled with red illustrates dyplasia; the cells have not yet progressed to cancer, but they are in danger of doing so, so they would be considered "pre-malignant." Dysplasia can revert back to hyperplasia, but it may also become malignant. Therefore, dysplasia should be carefully monitored or treated.
Dysplasia on a Biopsy from the Uterine Cervix
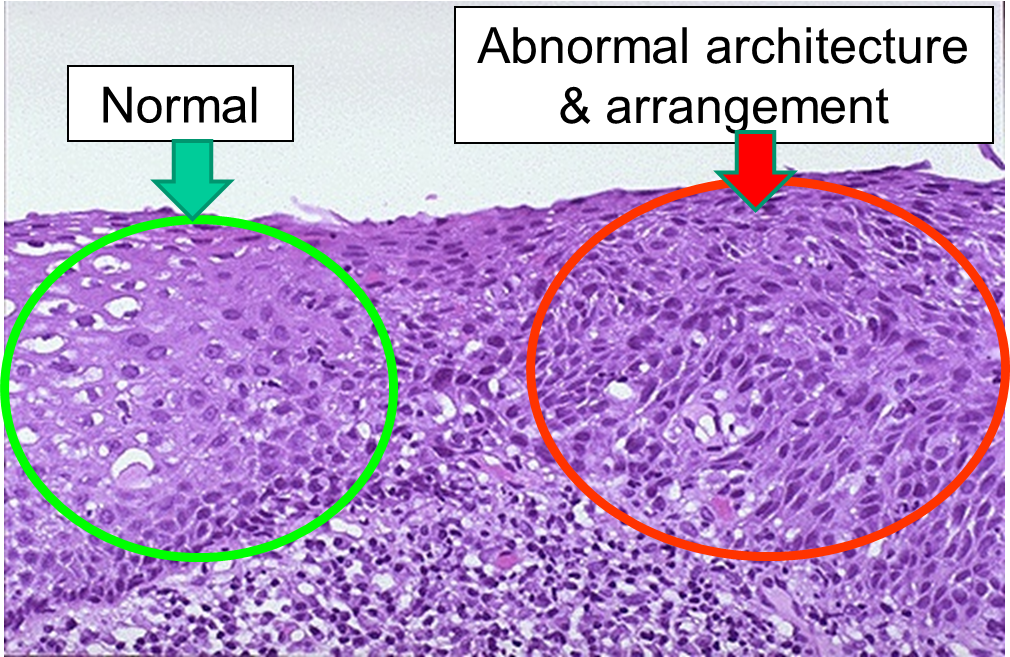
Image source: http://library.med.utah.edu/WebPath/FEMHTML/FEM008.html
Dysplasia on a Pap Smear
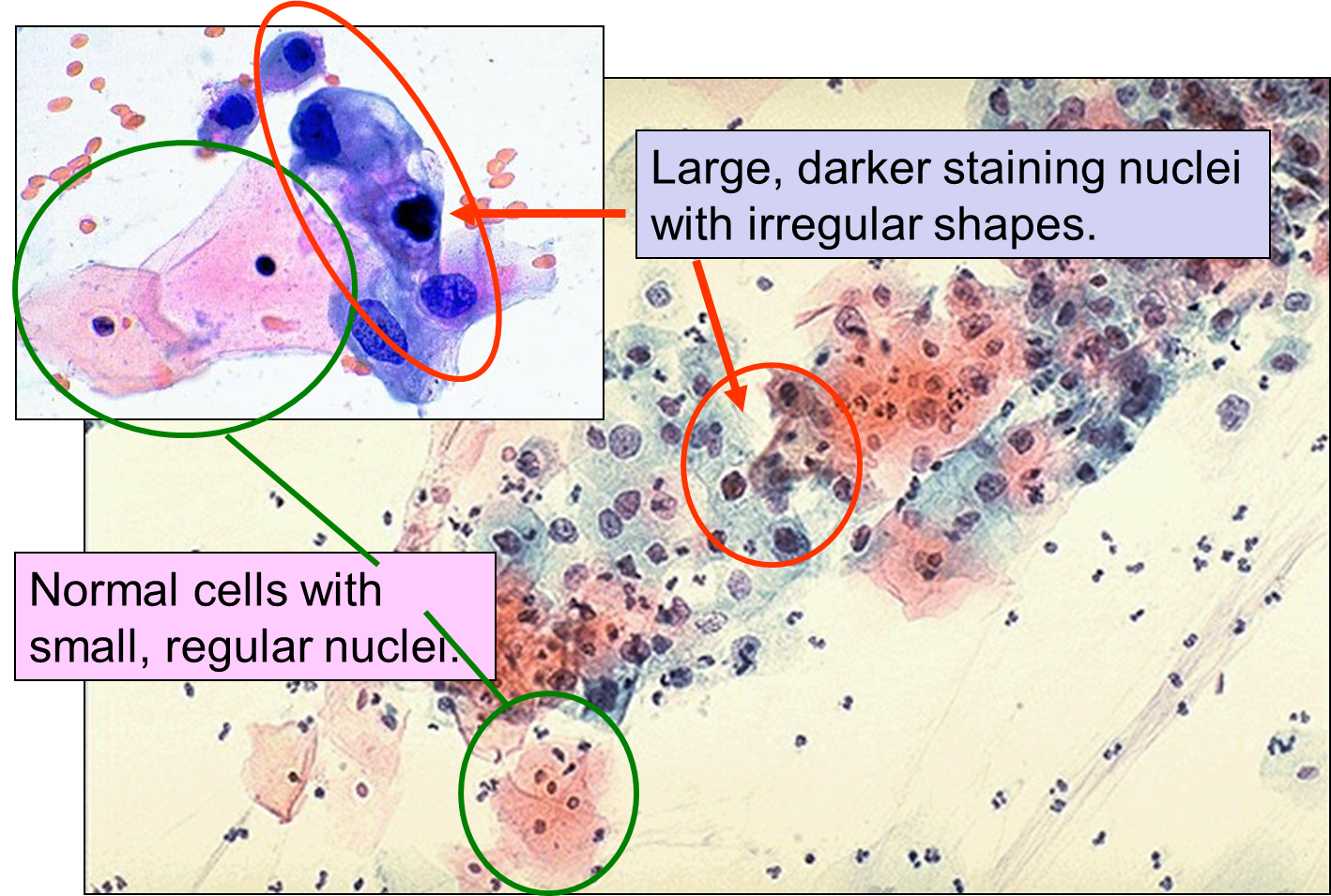
Carcinoma "in situ" literally means cancer in place. These cells have transitioned to being cancerous, but they have not yet invaded the adjacent tissues. Note that, in the illustration below, the in situ cancer is still confined to the epithelial layer from which it arose. If left untreated, the in situ cancer may remain confined to the epithelial layer indefinitely, but it may acquire additional mutations that enable it to progress to an invasive cancer..
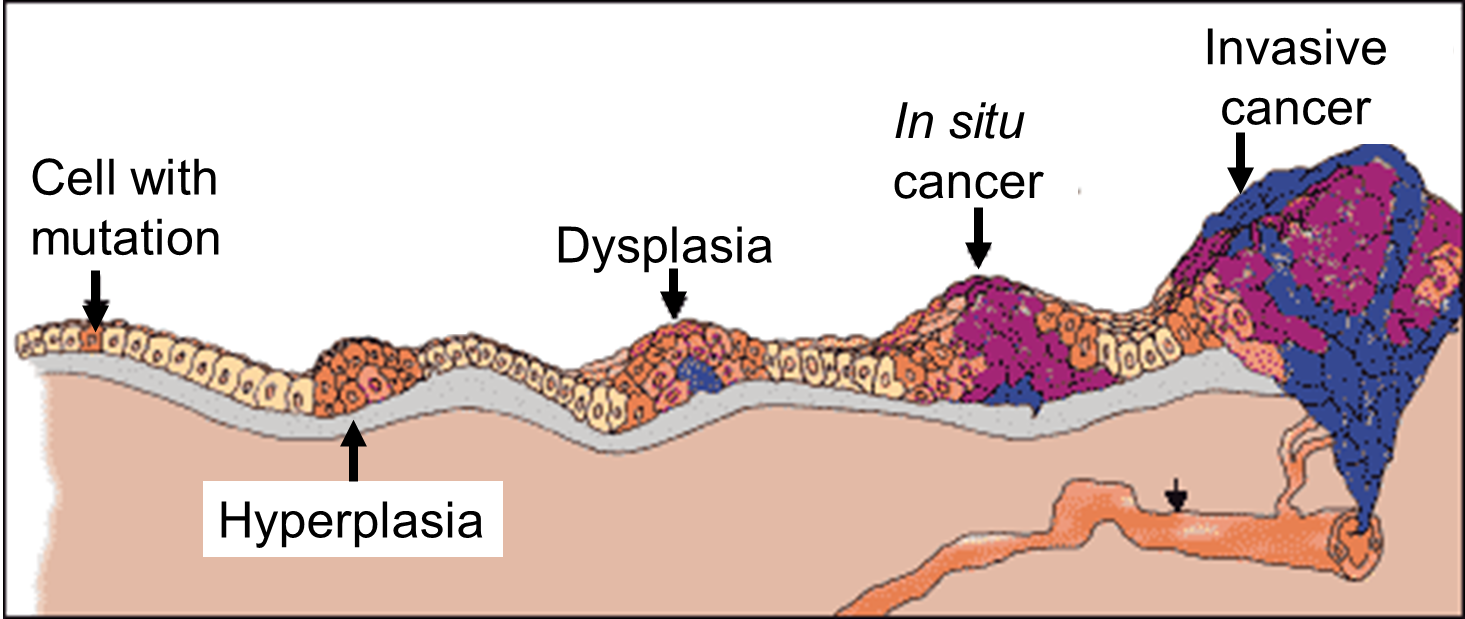
Source: http://www.ndhealthfacts.org/wiki/Oncology_%28Cancer%29
The progression begins with a mutation that makes the cell more likely to divide. The altered cell and its descendants grow and divide too often, a condition called hyperplasia. At some point, one of these cells experiences another mutation that further increases its tendency to divide; this cell's descendants divide excessively and look abnormal, a condition called dysplasia. As time passes, one of the cells experiences yet another mutation, causing very abnormal structure, loss of differentiation, and loss of contact between the cells; however, it is still confined to the epithelial layer from which it arose, so it is called a cancer in situ. The in situ cancer may remain contained indefinitely, but additional mutations may occur that enable it to invade neighboring tissues and shed cells into the blood or lymph, the tumor is said to be an invasive cancer (malignant). The escaped cells may establish new tumors (metastases) at other locations in the body. Source: http://science.education.nih.gov/supplements/nih1/cancer/guide/understanding1.htm
Metastasis is the movement or spreading of cancer cells from one organ or tissue to another and proliferate at the new site. Cancer cells usually spread through the bloodstream or the lymph system. The tumor mass can also spread locally, compress other structures, and damage surrounding tissues.
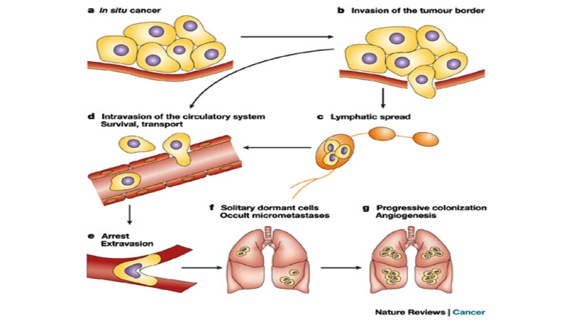
Source: http://www.nature.com/nrc/journal/v3/n1/fig_tab/nrc967_F1.html
The video below illustrates metastasis.
|
Excerpts from "The Emperor of All Maladies" by Siddhartha Mukherjee. "By the early 1990s, cancer biologists could begin to model the genesis of cancer in terms of molecular changes in genes. To understand that model, let us begin with a normal cell, say a lung cell that resides in the left lung of a forty-year old fire-safety-equipment installer. One morning in 1968, a minute sliver of asbestos from his equipment wafts through the air and lodges in the vicinity of that cell. His body reacts to the sliver with an inflammation. The cells around the sliver begin to divide furiously, like a minuscule wound trying to heal, and a small clump of cells derived from the original cell arises at the site. In one cell in that clump an accidental mutation occurs in the ras gene. The mutation creates an activated version of ras. The cell containing the mutant gene is driven to grow more swiftly than its neighbors and creates a clump within the original clump of cells. It is not yet a cancer cell, but a cell in which uncontrolled cell division has partly been unleashed - cancer's primordial ancestor. A decade passes. The small collection of ras-mutant cells continues to proliferate, unnoticed, in the far periphery of the lung. The man smokes cigarettes, and a carcinogenic chemical in tar reaches the periphery of the lung and collides with the clump of ras-mutated cells. A cell in this clump acquits a second mutation in its genes, activating a second oncogene. Another decade passes. Yet another cell in that secondary mass of cells is caught in the path of an errant X-ray and acquires yet another mutation, this time inactivating a tumor suppressor gene. This mutation has little effect since the cell possesses a second copy of that gene. But in the next year, another mutation inactivates the second copy of the tumor suppressor gene, creating a cell that possesses two activated oncogenes and an inactive tumor suppressor gene. Now a fatal march is on; an unraveling begins. The cells, now with four mutations, begin to outgrow their brethren. As the cells grow, they acquire additional mutations and they active pathways, resulting in cells even further adapted for growth and survival. One mutation in the tumor allows it to incite blood vessels to grow; another mutation within this blood-nourished tumor allows the tumor to survive even in areas of the body with low oxygen. Mutant cells beget cells. A gene that increases the mobility of the cells is activated in a cell. This cell, having acquired motility, can migrate through the lung tissue and enter the bloodstream. A descendant of this mobile cancer cell acquires the capacity to survive in the bone. This cell, having migrated through the blood, reaches the outer edge of the pelvis, where it begins yet another cycle of survival, selection, and colonization. It represents the first metastasis of a tumor that originated in the lung."
|
Cancer cells grow and divide at an abnormally rapid rate, are poorly differentiated, and have abnormal membranes, cytoskeletal proteins, and morphology. The abnormality in cells can be progressive with a slow transition from normal cells to benign tumors to malignant tumors.
In 2000 cancer biologists Robert Weinberg and Douglas Hanahan published an article entitled "The Hallmarks of Cancer." [Cell 2000;100(1):57-70] While they recognized that cancers occurred through a series of mutations in any of many genes. Despite this, they listed six essential alterations in cell physiology that characterized malignancy.
According to the American Cancer Society, there were approximately 11.4 million Americans with a history of cancer in 2010. Cancer is responsible for nearly 1 in every 4 deaths in the United States. In 2010, the mortality rate was 229.9 per 100,000 in males and 157.8 per 100,000 in females. The incidence rate was 556.5 per 100,000 in males and 414.8 per 100,000 in females. In the U.S., lung cancer is the most common cause of cancer mortality in men and women. (Source: American Cancer Society).
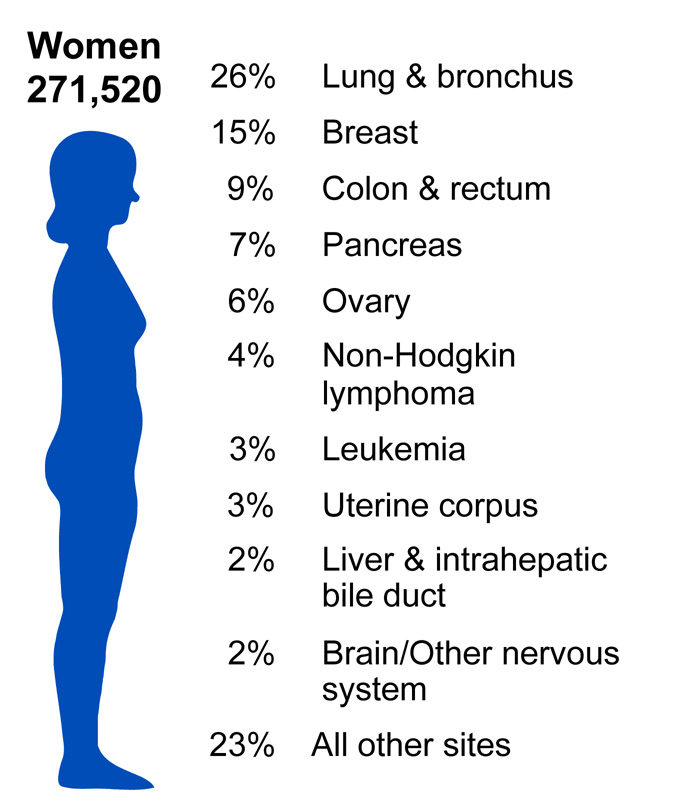
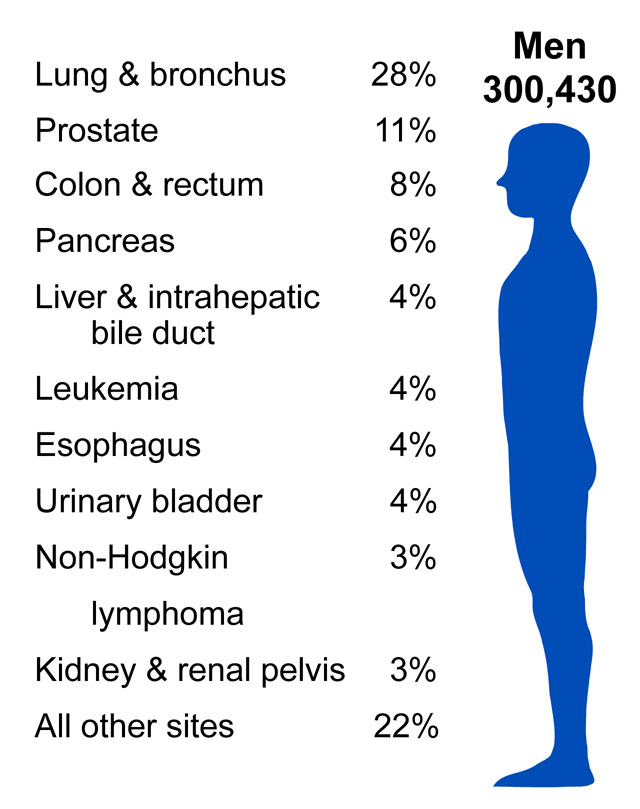
Source: http://www.cdc.gov/cancer/knowledge/provider-education/vaginal-vulvar/epidemiology.htm
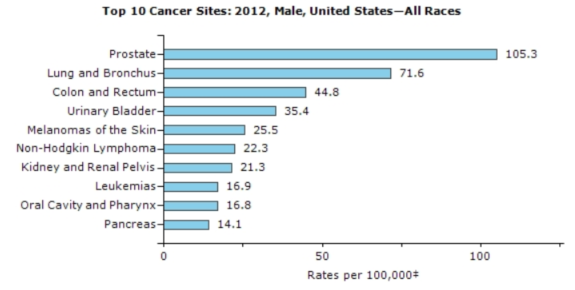
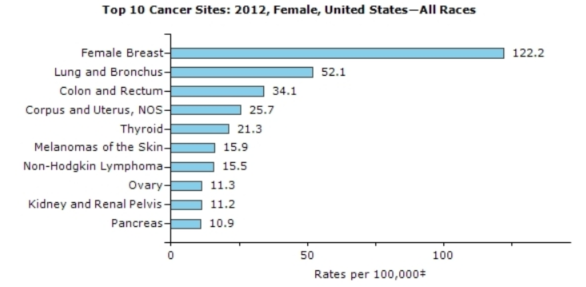
Source: https://nccd.cdc.gov/uscs/toptencancers.aspx
Benign tumors are abnormal growths that are no longer under normal regulation. They grow slowly, resemble normal cells, and are not cancerous. They grow only in one place and cannot spread or invade other parts of the body. They can however become harmful if they press on vital organs. Examples of benign tumors include skin moles, lipomas, hepatic adenomas.
These tumors are composed of embryonic, primitive, or poorly differentiated cells. They grow in a rapid, disorganized manner that is harmful to the body. They can also invade surrounding tissues and are become metastatic, initiating the growth of similar tumors in distant organs.
Cancers can be classified based on cell origin.
Cancer staging (illustrated on the right for colon cancer) describes the extent of a person's cancer based on:
For additional information see the National Cancer Institute web page on Cancer Staging.
The microscopic appearance a cancer indicates its likely behavior and its responsiveness to treatment.

Non-ionizing radiation is defined as a low frequency, long wavelength energy, which includes AM and FM radio signals, microwave ovens, power lines, heat lamps, visible light, and ultraviolet radiation.
Non-ionizing radiation is not energetic enough to penetrate deeply or to create free radicals. It only penetrates single-celled organisms and the superficial cell layers of multicellular organisms. Its energy is sufficient to boost the energy of electrons in molecules. Nucleotides in DNA absorb this energy (maximum mutagenesis occurs at 234 nm). Excitation of nucleotides can induce abnormalities such as formation of bulky thymine dimers that cause mutations by interfering with proper base pairing during replication.
Ultraviolet radiation from sun exposure and tanning salons is a major cause of skin cancer, accounting for 50-90% of all skin cancers, according to the World Health Organization. Ultraviolet radiation penetrates the superficial layers of the skin and causes mutations when the radiant energy is absorbed and disrupts bonds within DNA. Ultraviolet light also causes premature aging of skin and eye damage, including cataracts. For more information on the health effects of ultraviolet radiation, see the module on Sun Exposure.
The National Institutes of Health (NIH) has a good web site providing an array of information about sun damage and skin cancer. See NIH Senior Health.
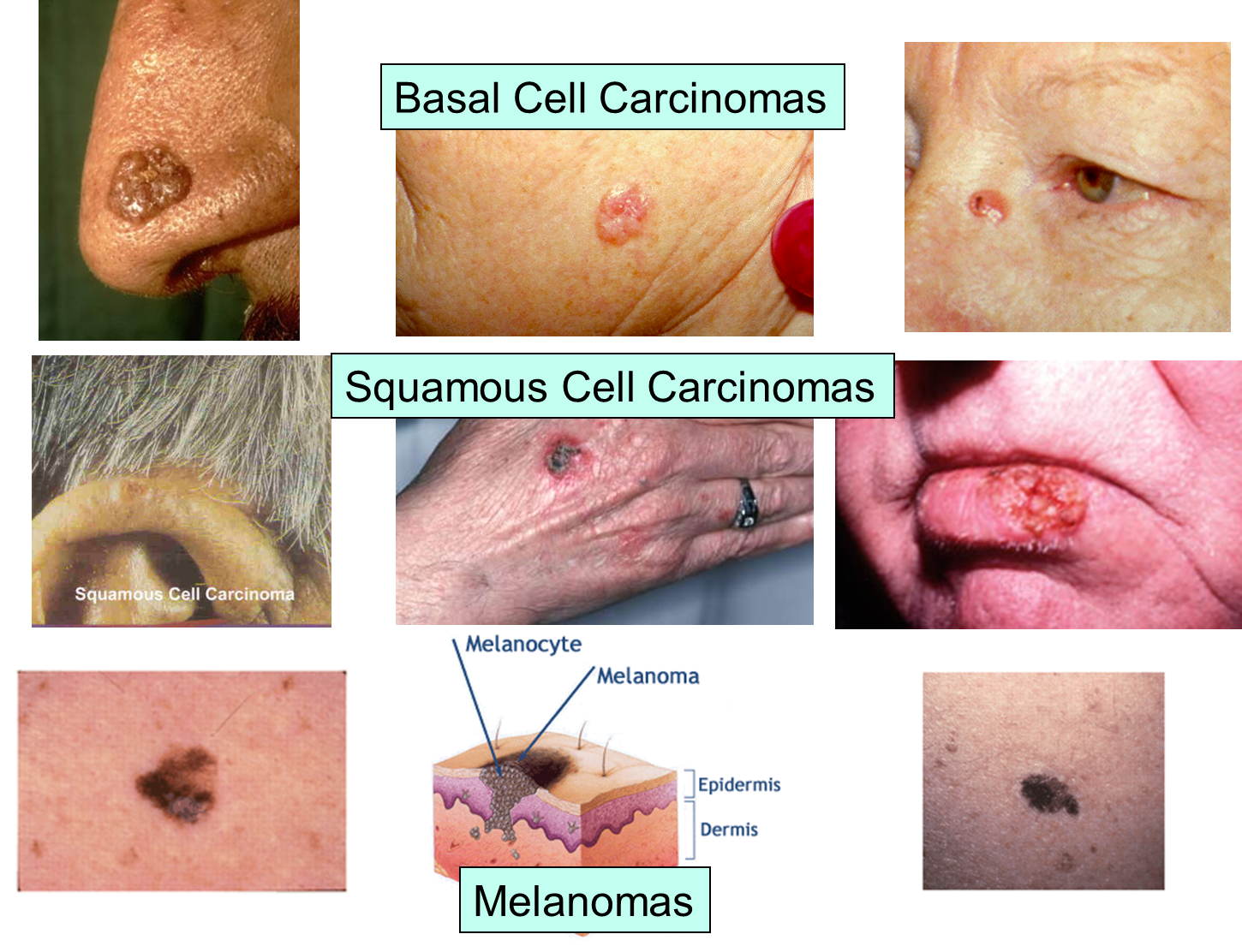
The images below illustrate the appearance of the three major forms of skin cancer.
The next set of images illustrate the characteristics of melanoma, and the changes that occur during its evolution.
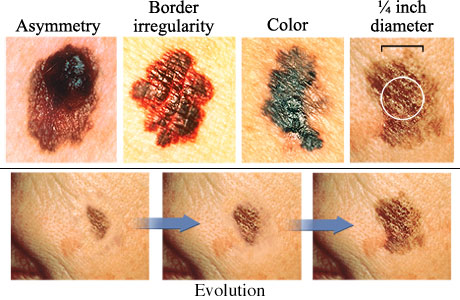
Source: http://www.webmd.com/melanoma-skin-cancer/abcds-of-melanoma-skin-cancer
|
Do cell phones cause cancer? From National Cancer Institute:
|
Protons, neutrons, x-rays, & gamma rays have highly energetic wavelengths, frequency which penetrate deeply into tissues. Lead shields block these forms of radiation.
Ionizing radiation can damage DNA with a direct hit, but it more frequently damages DNA indirectly by stripping away electrons when they strike a molecule, thereby creating a highly reactive free radicals. The resulting free radicals can damage other molecules by stealing their electrons, and they also can break phosphate bonds in DNA. Breaks in a single strand can be repaired easily, but sometimes double strand breaks are not. Ionizing radiation is very dangerous because its high energy allows it to penetrate deeply into tissue leaving a trail of free radicals in its path. Ionizing radiation has a cumulative mutagenic effect.
Examples:
A chemical carcinogen is any discrete chemical compound, which has been shown to cause cancer. There are several classes of chemicals that tend to be carcinogenic. Within these classes, some commonly encountered chemicals include formaldehyde, chloroform, asbestos, arsenic, and aflatoxin. Carcinogens may enter the body through skin absorption, ingestion, or inhalation attacking many organs. Dose, or the amount and length of exposure, directly affect the amount of damage obtained. To reduce exposure to these common carcinogens use proper ventilation and hygienic habits.
Tobacco use accounts for at least 30% of cancer deaths and 87% of lung cancer deaths. Tobacco instigates cancer of the lung, oral cavity, nasal cavity, larynx, oropharynx, hypopharynx, esophagus, stomach, liver, bladder, ureter, kidney, cervix, and myeloid leukemia. Interestingly enough, lung cancer is the most preventable form of cancer in our society today. Cigarette smoke contains more than 60 carcinogens. Therefore, the cessation of cigarette smoking would immensely decrease many cancer occurrences.
For more information on chemical carcinogens, please see:
http://www.cancer.org/Cancer/CancerCauses/OtherCarcinogens/GeneralInformationaboutCarcinogens/known-and-probable-human-carcinogens
Heterocyclic amines (HCAs) and polycyclic aromatic hydrocarbons (PAHs) are a family of carcinogenic chemicals formed from the cooking of muscle meats (beef, pork, fowl, and fish); other sources of protein have not been associated with HCAs (e.g. eggs, tofu). HCAs form when amino acids and creatine (a chemical found in muscles) react at high cooking temperatures. Frying, broiling, and barbecuing produce the largest amounts of HCAs because the meats are cooked at very high temperatures. They have been associated with an increased risk of stomach, colorectal, and pancreatic cancer.
For more information, see http://www.cancer.gov/cancertopics/factsheet/Risk/cooked-meats.
Viruses are estimated to cause 15 – 20% of all cancers in humans. Some factors that influence the progression to cancer development include the host's genetic susceptibility, mutations, exposure to other carcinogens, and immune system deficiencies. Oncoviruses include the Epstein –Barr virus, hepatitis B virus, human papilloma virus (HPV), herpes virus, and hepatitis C virus.
Although most strains of HPV will not remain active in the body for more than several years, a number of aggressive strains are significantly more likely to cause cancer. These strains activate certain oncogenes. When these oncogenes impede factors that limit cell growth, cells divide much more rapidly than normal, and can lead to tumor development.
Chronic infection with hepatitis causes 80% of all primary liver cancers worldwide. The most common risk factor for liver cancer is chronic infection with hepatitis B virus (HBV). HBV attacks the liver, leading to progressive liver damage, which in turn develops into liver cancer. Those infected with HPV are 100 times more likely to develop liver cancers than those who are not infected. There is an effective vaccine against HBV. There are effective therapies to manage chronic hepatitis B infections and help prevent progression to liver cancer.
For further information on hepatitis B, consult the following module: http://www.hepb.org/hepatitisbcd/modules/infectd/id450101/id459101g.html
AIDS is not a direct cause of cancer, but it indirectly increases the risk by impairing immune function. The National Cancer Institute states the following:
"Definition of AIDS-related cancers: Certain cancer types that are more likely to occur in people who are infected with the human immunodeficiency virus (HIV). The most common types are Kaposi sarcoma and non-Hodgkin lymphoma. Other AIDS-related cancers include Hodgkin disease and cancers of the lung, mouth, cervix, and digestive system."
People with AIDS are 310 times more likely to develop Kaposi's sarcoma and 113 times more likely to develop non-Hodgkin's lymphoma (Goedert, J. J., Coté, T. R., Virgo, P., Scoppa, S. M., Kingma, D. W., Gail, M. H., Biggar, R. J. (1998). Spectrum of AIDS-associated malignant disorders. The Lancet, 351(9119), 1833-1839. doi:10.1016/S0140-6736(97)09028-4.)
High body mass index is associated with an increased risk of colon, breast, endometrium, kidney, esophagus, gastric cardia, pancreas, gallbladder and liver cancer.
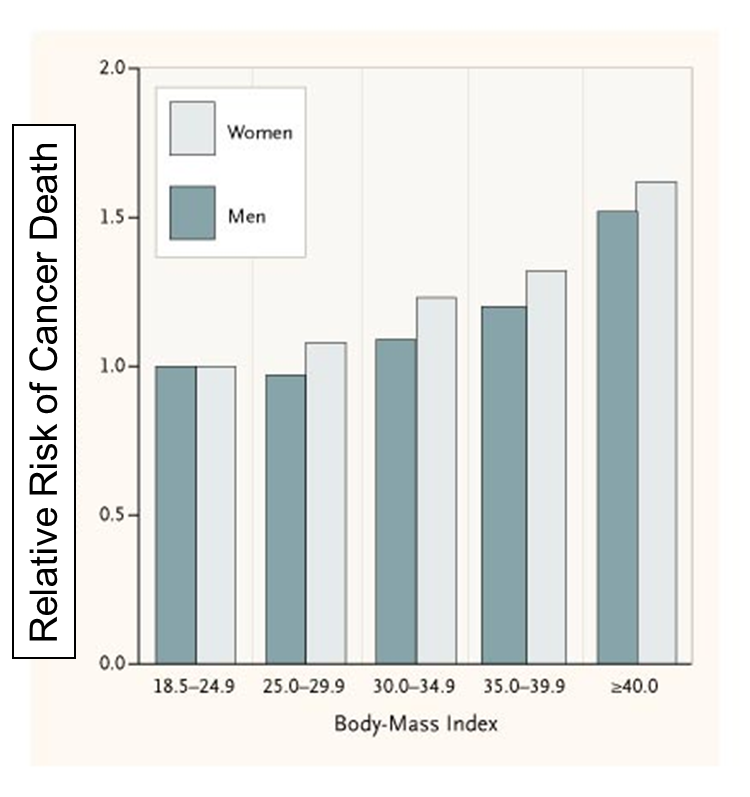
The National Cancer Institute says the following regarding the possible mechanisms for this association:
"Several possible mechanisms have been suggested to explain the association of obesity with increased risk of certain cancers:
Limiting fat and calorie intake may decrease risk of some cancers (e.g., breast and colon cancer). In addition, higher consumption of fruits & vegetables is associated with a reduction in cancer risk. The responsible components remain unknown, but many recommend at least 5 servings/day.
Angiogenesis is the formation of new blood vessels. This normally takes place during growth and during healing after a tissue injury. Tumors also need new blood vessels in order to grow, and there is now abundant evidence that inhibitors of angiogenesis can stop or at least slow the growth of tumors. Several angiogenesis inhibitors have been approved for use in cancer treatment by the Food and Drug Administration. (For more information see the following link from the National Institutes of Health:
http://www.cancer.gov/cancertopics/factsheet/Therapy/angiogenesis-inhibitorsIn addition, there is also evidence that there are a number of naturally occurring angiogenesis inhibitors in some vegetables and fruits. To learn more about these, watch the video below from TED.com.
Alcohol intake is associated with an increased risk of cancer of the mouth, esophagus, pharynx and larynx, colon, and liver. Consumption of alcohol is the primary cause of liver cancer; oral and pharyngeal cancers are more common in alcohol users than in non-alcohol users. Smokers who also drink are at an increased risk of developing cancer. According to the American Cancer Society, drinking in moderation is key to reducing the risk of alcohol-related cancers; men should have no more than two drinks per day and women should have no more than one.
Most cancers are sporadic, i.e., with no hereditary predisposition. However, a predisposition to certain types of cancers can be hereditary. Some examples are:
For more information on heredity and cancer, refer to: http://www.cancer.org/Cancer/CancerCauses/GeneticsandCancer/heredity-and-cancer
For an excellent and succinct summary of risk factors for breast cancer, see http://www.cancer.org/cancer/breastcancer/detailedguide/breast-cancer-risk-factors
The National Cancer Society also provides an online breast cancer risk calculator.
As noted above, the BRCA1 and BRCA2 mutations can be inherited and lead to an increased risk of breast and other types of cancer. The National Cancer Institute Fact Sheet on BRCA1 and BRCA2 states:
"A woman's lifetime risk of developing breast and/or ovarian cancer is greatly increased if she inherits a harmful mutation in BRCA1 or BRCA2. Such a woman has an increased risk of developing breast and/or ovarian cancer at an early age (before menopause) and often has multiple, close family members who have been diagnosed with these diseases. Harmful BRCA1 mutations may also increase a woman's risk of developing cervical, uterine, pancreatic, and colon cancer (1, 2). Harmful BRCA2 mutations may additionally increase the risk of pancreatic cancer, stomach cancer, gallbladder and bile duct cancer, and melanoma (3).
Men with harmful BRCA1 mutations also have an increased risk of breast cancer and, possibly, of pancreatic cancer, testicular cancer, and early-onset prostate cancer. However, male breast cancer, pancreatic cancer, and prostate cancer appear to be more strongly associated with BRCA2 gene mutations (2–4).
The likelihood that a breast and/or ovarian cancer is associated with a harmful mutation in BRCA1 or BRCA2 is highest in families with a history of multiple cases of breast cancer, cases of both breast and ovarian cancer, one or more family members with two primary cancers (original tumors that develop at different sites in the body), or an Ashkenazi (Central and Eastern European) Jewish background. However, not every woman in such families carries a harmful BRCA1 or BRCA2 mutation, and not every cancer in such families is linked to a harmful mutation in one of these genes. Furthermore, not every woman who has a harmful BRCA1 or BRCA2 mutation will develop breast and/or ovarian cancer."
Nevertheless, it is estimated that 85% of breast cancers are sporadic, i.e., not do to an inherited mutation.
A woman's risk of developing breast cancer depends on hormonal and reproductive history. In essence, any factor that increases a women's total lifetime exposure to estrogens (either endogenous or exogenous) increases her risk. Estrogens stimulate growth of breast tissue and appear to have a role in the development & growth of breast cancer. Estrogens promote the development of mammary cancer in rodents. They also stimulate proliferation of human breast cancer cells grown in cell culture.
Factors that would contribute to lifetime exposure to endogenous estrogens would include:
Exogenous Sources of Estrogen (from an external source, i.e., medications and birth control)
|
Oral Contraceptives and Cancer From National Cancer Institute: "A 1996 analysis of epidemiologic data from more than 50 studies worldwide by the Collaborative Group on Hormonal Factors in Breast Cancer found that women who were current or recent users of birth control pills had a slightly higher risk of developing breast cancer than women who had never used the pill (2). The risk was highest for women who started using oral contraceptives as teenagers. However, 10 or more years after women stopped using oral contraceptives, their risk of developing breast cancer had returned to the same level as if they had never used birth control pills...."
http://www.cancer.gov/cancertopics/factsheet/Risk/oral-contraceptives http://jnci.oxfordjournals.org/content/94/23/1773.long
|
|
Tamoxifen - An Estrogen Blocking Drug Tamoxifen is a drug that has an estrogen-blocking effect in the breast, but estrogen-like effects on some other tissues, e.g. uterus & bone.
The Breast Cancer Prevention Trial studied effect of Tamoxifen for 5 years in a large group of women at increased risk. Women ages 35 and older who took a daily dose of 20 mg Tamoxifen for up to 5 years had 50% reduction in risk of breast cancer.
|
In the video clip below, Dr. David Sherr, from the environmental health department at BUSPH, discusses the potential role of environmental pollutants as risk factors for breast (and other) cancers. Dr. Sherr's research is exploring the molecular mechanisms that initiate and maintain breast cancer. Much of his research has focused on the role of an environmental chemical receptor (the aryl hydrocarbon receptor - AhR) and the role of polycyclic aromatic hydrocarbons (PAHs), which are atmospheric pollutants generated by burning fuel and by cooking meat at high temperatures (e.g., grilling). PAHs are also found in smoked fish. In this video, Dr. Sherr also discusses the difficulties in establishing a causal link between environmental pollutants and cancer.
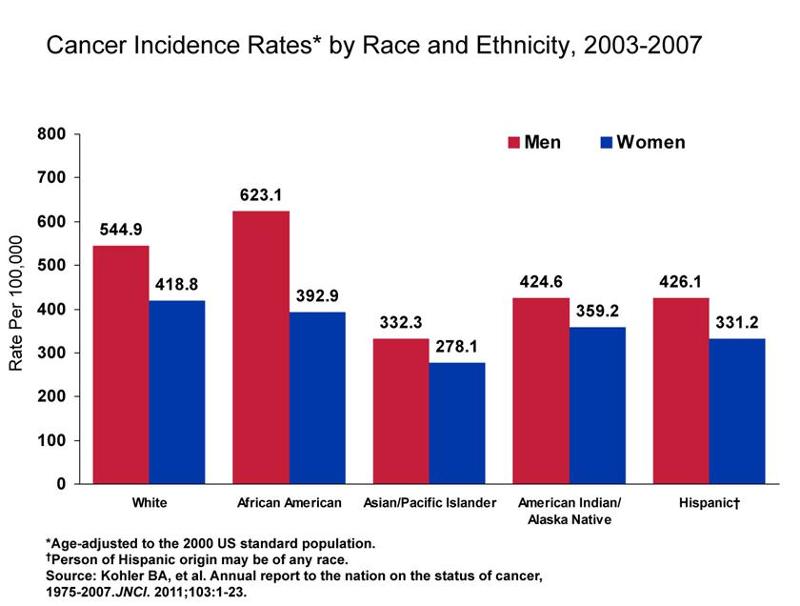
There are varying rates of cancer incidence and mortality among the different races and socioeconomic statuses. As shown in the graph below, African American men continue to have a disproportionately higher incidence of cancer.
Other disparities are outlined below:
There are complex, interrelated factors that contribute to health disparities regarding cancer. However, the two most common factors seem to be a lack of health care coverage and low socioeconomic status (SES). According to the National Cancer Institute, SES is a stronger predictor of cancer risk and risk of cancer death than race. Access to health care affects the stage at which a person is diagnosed with cancer and living conditions determine whether a person is exposed to certain environmental toxins that can increase the risk of cancer. SES also appears to play a major role in influencing the prevalence of behavioral risk factors for cancer. This includes smoking, lack of physical activity, obesity, and excessive alcohol consumption. African Americans generally have a lower SES and subsequently less access to health care, which may lead to the disparities in cancer incidence and mortality rates.

Do you know myth from fact?
Are the following statements true, or are they myths? Decide what you think and then mouse over the statement to find the answer.
Other common cancer myths dispelled: http://www.mayoclinic.com/health/cancer/HO00033